|
|
| [Code of Federal Regulations] |
| [Title 21, Volume 4] |
| [CITE: 21CFR201] |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES
SUBCHAPTER C - DRUGS: GENERAL
|
|
Subpart G - Specific Labeling Requirements for Specific Drug Products
|
|
Sec. 201.300 Notice to manufacturers, packers, and distributors of glandular preparations.
|
|
(a) Under date of December 4, 1941, in a notice to manufacturers of glandular preparations, the Food and Drug Administration expressed the opinion that preparations of inert glandular materials intended for medicinal use should, in view of the requirement of section 201(n) of the Federal Food, Drug, and Cosmetic Act (52 Stat. 1041; 21 U.S.C. 321(n)), be labeled with a statement of the material fact that there is no scientific evidence that the articles contain any therapeutic or physiologically active constituents. Numerous preparations of such inert glandular materials were subsequently marketed with disclaimers of the type suggested. The term inert glandular materials means preparations incapable of exerting an action or effect of some significant or measurable benefit in one way or another, i.e., in the diagnosis, cure, mitigation, treatment, or prevention of disease, or in affecting the structure or any function of the body.
(b) Manufacturers have heretofore taken advantage of § 201.100 permitting omission of directions for use when the label bears the prescription legend. Section 201.100(c) requires that the labeling of the drug, which may include brochures readily available to licensed practitioners, bear information as to the use of the drug by practitioners licensed by law to administer it. Obviously, information adequate for the use of an inert glandular preparation is not available to practitioners licensed by law.
(c) The Department of Health and Human Services is of the opinion that inert glandular materials may not be exempted from the requirements of section 502(f)(1) of the act that they bear adequate directions for use; and, accordingly, that their labeling must include among other things, representations as to the conditions for which such articles are intended to be used or as to the structure or function of the human body that they are intended to affect. Since any such representations offering these articles for use as drugs would be false or misleading, such articles will be considered to be misbranded if they are distributed for use as drugs.
(d) The amended regulations provide also that in the case of drugs intended for parenteral administration there shall be no exemption from the requirement that their labelings bear adequate directions for use. Such inert glandular materials for parenteral use are therefore subject to the same comment as applies to those intended for oral administration.
|
|
|
Sec. 201.301 Notice to manufacturers, packers, and distributors of estrogenic hormone preparations.
|
|
Some drug preparations fabricated wholly or in part from estradiol and labeled as to potency in terms of international units or in terms of international units of estrone activity have been marketed. The international unit of the estrus-producing hormone was established by the International Conference on the Standardization of Sex Hormones at London, England, on August 1, 1932. This unit was defined as "the specific estrus-producing activity contained in 0.1 gamma ( = 0.0001 mg.) of the standard" hydroxyketonic hormone found in urine (estrone). The International Conference declared that it did not recommend the determination of the activity of nonhydroxyketonic forms of estrogenic hormones in units of estrone because of the varying ratios between the activity of such nonhydroxyketonic estrogenic hormones and estrone, when measured by different methods on test animals. There is no international unit for measuring the activity of estradiol and no accepted relationship between its activity and that of estrone, either in test animals or in humans. The declaration of potency of estradiol in terms of international units or in terms of international units of estrone activity is therefore considered misleading, within the meaning of 21 U.S.C. 352(a). The declaration of the estradiol content of an estrogenic hormone preparation in terms of weight is considered appropriate.
|
|
|
Sec. 201.302 Notice to manufacturers, packers, and distributors of drugs for internal use which contain mineral oil.
|
|
(a) In the past few years research studies have altered medical opinion as to the usefulness and harmfulness of mineral oil in the human body. These studies have indicated that when mineral oil is used orally near mealtime it interferes with absorption from the digestive tract of provitamin A and the fat-soluble vitamins A, D, and K, and consequently interferes with the utilization of calcium and phosphorus, with the result that the user is left liable to deficiency diseases. When so used in pregnancy it predisposes to hemorrhagic disease of the newborn.
(b) There is accumulated evidence that the indiscriminate administration of mineral oil to infants may be followed by aspiration of the mineral oil and subsequent "lipoid pneumonia."
(c) In view of these facts, the Department of Health and Human Services will regard as misbranded under the provisions of the Federal Food, Drug, and Cosmetic Act a drug for oral administration consisting in whole or in part of mineral oil, the labeling of which encourages its use in pregnancy or indicates or implies that such drug is for administration to infants.
(d) It is also this Department's view that the act requires the labelings of such drugs to bear a warning against consumption other than at bedtime and against administration to infants. The following form of warning is suggested: "Caution: To be taken only at bedtime. Do not use at any other time or administer to infants, except upon the advice of a physician."
(e) This statement of interpretation does not in any way exempt mineral oil or preparations containing mineral oil from complying in all other respects with the requirements of the Federal Food, Drug, and Cosmetic Act.
|
|
|
Sec. 201.303 Labeling of drug preparations containing significant proportions of wintergreen oil.
|
|
(a) Because methyl salicylate (wintergreen oil) manifests no toxicity in the minute amounts in which it is used as a flavoring, it is mistakenly regarded by the public as harmless even when taken in substantially larger amounts. Actually, it is quite toxic when taken in quantities of a teaspoonful or more. Wintergreen oil and preparations containing it have caused a number of deaths through accidental misuse by both adults and children. Children are particularly attracted by the odor and are likely to swallow these products when left within reach.
(b) To safeguard against fatalities from this cause, the Department of Health and Human Services will regard as misbranded under the provisions of the Federal Food, Drug, and Cosmetic Act any drug containing more than 5 percent methyl salicylate (wintergreen oil), the labeling of which fails to warn that use otherwise than as directed therein may be dangerous and that the article should be kept out of reach of children to prevent accidental poisoning.
(c) This statement of interpretation in no way exempts methyl salicylate (wintergreen oil) or its preparations from complying in all other respects with the requirements of the Federal Food, Drug, and Cosmetic Act.
|
|
|
Sec. 201.304 Tannic acid and barium enema preparations.
|
|
(a) It has become a widespread practice for tannic acid to be added to barium enemas to improve X-ray pictures. Tannic acid is capable of causing diminished liver function and severe liver necrosis when absorbed in sufficient amounts. The medical literature reports a number of deaths associated with the addition of tannic acid to barium enemas. There is a lack of scientific evidence to establish the conditions, if any, under which tannic acid is safe and effective for use in enemas. Tannic acid for rectal use to enhance X-ray visualization is regarded as a new drug within the meaning of section 201(p) of the Federal Food, Drug, and Cosmetic Act.
(b) In view of the hazards involved when tannic acid is used in barium enemas, any shipments of tannic acid labeled to come within the exemptions under 502(f) of the Act containing such phrases as: "Caution: For manufacturing, processing, or repackaging," "For prescription compounding," or "Diagnostic reagent - For professional use only" will be regarded by the Commissioner of Food and Drugs as misbranded within the meaning of section 502(f) of the Federal Food, Drug, and Cosmetic Act unless the label and the labeling bear conspicuously a warning to the effect: "Warning - Not for use in enemas."
(c) Any tannic acid intended for use by man and found within the jurisdiction of the Federal Food, Drug, and Cosmetic Act labeled contrary to this section after 60 days from the date of its publication in the Federal Register may be made the subject of regulatory proceedings.
|
|
|
Sec. 201.305 Isoproterenol inhalation preparations (pressurized aerosols, nebulizers, powders) for human use; warnings.
|
|
(a) Accumulating reports have been received by the Food and Drug Administration and have appeared in the medical literature of severe paradoxical bronchoconstriction associated with repeated, excessive use of isoproterenol inhalation preparations in the treatment of bronchial asthma and other chronic bronchopulmonary disorders. The cause of this paradoxical reaction is unknown; it has been observed, however, that patients have not responded completely to other forms of therapy until use of the isoproterenol inhalation preparation was discontinued. In addition, sudden unexpected deaths have been associated with the excessive use of isoproterenol inhalation preparations. The mechanism of these deaths and their relationship, if any, to the cases of severe paradoxical bronchospasm are not clear. Cardiac arrest was noted in several of these cases of sudden death.
(b) On the basis of the above information and after discussion with and concurrence of the Respiratory and Anesthetic Drugs Advisory Committee for Food and Drug Administration, the Commissioner of Food and Drugs concludes that in order for the labeling of such drugs to bear adequate information for their safe use, as required by § 201.100, such labeling must include the following:
Warning: Occasional patients have been reported to develop severe paradoxical airway resistance with repeated, excessive use of isoproterenol inhalation preparations. The cause of this refractory state is unknown. It is advisable that in such instances the use of this preparation be discontinued immediately and alternative therapy instituted, since in the reported cases the patients did not respond to other forms of therapy until the drug was withdrawn.
Deaths have been reported following excessive use of isoproterenol inhalation preparations and the exact cause is unknown. Cardiac arrest was noted in several instances.
(c)(1) The Commissioner also concludes that in view of the manner in which these preparations are self-administered for relief of attacks of bronchial asthma and other chronic bronchopulmonary disorders, it is necessary for the protection of users that warning information to patients be included as a part of the label and as part of any instructions to patients included in the package dispensed to the patient as follows:
Warning: Do not exceed the dose prescribed by your physician. If difficulty in breathing persists, contact your physician immediately.
(2) The warning on the label may be accomplished (i) by including it on the immediate container label with a statement directed to pharmacists not to remove the label or (ii) by including in the package a printed warning with instructions to pharmacists to place the warning on the container prior to dispensing.
(d) The marketing of isoproterenol inhalation preparations may be continued if all the following conditions are met:
(1) Within 30 days following the date of publication of this section in the Federal Register:
(i) The label and labeling of such preparations shipped within the jurisdiction of the act are in accordance with paragraphs (b) and (c) of this section.
(ii) The holder of an approved new-drug application for such preparation submits a supplement to his new-drug application to provide for appropriate labeling changes as described in paragraphs (b) and (c) of this section.
(2) Within 90 days following the date of publication of this section in the Federal Register, the manufacturer, packer, or distributor of any drug containing isoproterenol intended for inhalation for which a new-drug approval is not in effect submits a new-drug application containing satisfactory information of the kinds required by § 314.50 of this chapter, including appropriate labeling as described in paragraphs (b) and (c) of this section.
(3) The applicant submits additional information required for the approval of the application as may be specified in a written communication from the Food and Drug Administration.
(e) After 270 days following expiration of said 90 days, regulatory proceedings based on section 505(a) of the Federal Food, Drug, and Cosmetic Act may be initiated with regard to any such drug shipped within the jurisdiction of the act for which an approved new-drug application is not in effect.
[40 FR 13998, Mar. 27, 1975, as amended at 55 FR 11576, Mar. 29, 1990]
|
|
|
Sec. 201.306 Potassium salt preparations intended for oral ingestion by man.
|
|
(a) The Food and Drug Administration will initiate no regulatory action with respect to the continued marketing of coated tablets containing potassium chloride or other potassium salts which supply 100 milligrams or more of potassium per tablet provided all the following conditions are met:
(1) Within 30 days from the date of publication of this statement of policy in the Federal Register:
(i) The labeling of the drug bears the prescription caution statement quoted in section 503(b)(4) of the Federal Food, Drug, and Cosmetic Act;
(ii) The labeling on or within the package from which the drug is to be dispensed bears adequate information for its use by practitioners in accord with the "full disclosure" labeling requirements of § 201.100 of this chapter, including the following warning statement:
Warning - There have been several reports, published and unpublished, concerning nonspecific small-bowel lesions consisting of stenosis, with or without ulceration, associated with the administration of enteric-coated thiazides with potassium salts. These lesions may occur with enteric-coated potassium tablets alone or when they are used with nonenteric-coated thiazides, or certain other oral diuretics. These small-bowel lesions have caused obstruction, hemorrhage, and perforation. Surgery was frequently required and deaths have occurred. Based on a large survey of physicians and hospitals, both United States and foreign, the incidence of these lesions is low, and a causal relationship in man has not been definitely established. Available information tends to implicate enteric-coated potassium salts, although lesions of this type also occur spontaneously. Therefore, coated potassium-containing formulations should be administered only when indicated, and should be discontinued immediately if abdominal pain, distention, nausea, vomiting, or gastrointestinal bleeding occur. Coated potassium tablets should be used only when adequate dietary supplementation is not practicable.
(Although the warning statement includes references to enteric-coated potassium salt preparations, it applies to any capsule or coated tablet of a potassium salt intended for oral ingestion without prior dilution with an adequate volume of liquid to preclude gastrointestinal injury.)
(iii) Any other labeling or additional advertising for the drug conforms to the labeling described in paragraph (a)(1)(ii) of this section, in accordance with §§ 202.1 and 201.100 of this chapter.
(2) Within 90 days from the date of publication of this statement of policy in the Federal Register, the manufacturer, packer, or distributor of the drug shall submit a new-drug application containing satisfactory information of the kind required by § 314.50 of this chapter, with appropriate labeling as described in this paragraph.
(b) The Food and Drug Administration may initiate regulatory proceedings after 30 days from the date of publication of this section, with respect to the marketing of uncoated tablets containing potassium chloride or other potassium salts which supply 100 milligrams or more of potassium per tablet or with respect to liquid preparations containing potassium chloride or other potassium salts which supply 20 milligrams or more of potassium per milliliter, labeled or intended for human use, unless all the following conditions are met:
(1) The labeling of the drug bears the prescription statement quoted in section 503(b)(4) of the Federal Food, Drug, and Cosmetic Act; and
(2) The labeling on or within the package from which the drug is to be dispensed bears adequate information for its use by practitioners in accord with the "full disclosure" labeling requirements of § 201.100 of this chapter, including a recommendation that patients be directed to dissolve any such tablets in an appropriate amount of liquid and to dilute any such liquid preparations adequately to assure against gastrointestinal injury associated with the oral ingestion of concentrated potassium salt preparations.
[40 FR 13998, Mar. 27, 1975, as amended at 55 FR 11576, Mar. 29, 1990; 67 FR 4906, Feb. 1, 2002]
|
|
|
Sec. 201.307 Sodium phosphates; package size limitation, warnings, and directions for over-the-counter sale.
|
|
(a) Reports in the medical literature and data accumulated by the Food and Drug Administration indicate that multiple container sizes of sodium phosphates oral solution available in the marketplace have caused consumer confusion and appear to have been involved in several consumer deaths. Sodium phosphates oral solution has been marketed in 45-milliliter (mL), 90-mL, and 240-mL container sizes. The 45-mL and 90-mL container sizes of sodium phosphates oral solution are often recommended and prescribed by physicians for bowel cleansing prior to surgery and diagnostic procedures of the colon. Sodium phosphates oral solution (adult dose 20 mL to 45 mL) is also used as an over-the-counter (OTC) laxative for the relief of occasional constipation. Accidental overdosing and deaths have occurred because the 240-mL container was mistakenly used instead of the 45-mL or 90-mL container. The Food and Drug Administration is limiting the amount of sodium phosphates oral solution to not more than 90 mL (3 ounces (oz)) per OTC container because of the serious health risks associated with the ingestion of larger than intended doses of this product. Further, because an overdose of either oral or rectal enema sodium phosphates can cause an electrolyte imbalance, additional warning and direction statements are required for the safe use of any OTC laxative drug product containing sodium phosphates.
(b) Any OTC drug product for laxative or bowel cleansing use containing sodium phosphates as an active ingredient when marketed as described in paragraph (a) of this section is misbranded within the meaning of section 502 of the Federal Food, Drug, and Cosmetic Act unless packaged and labeled as follows:
(1) Package size limitation for sodium phosphates oral solution: Container shall not contain more than 90 mL (3 oz).
(2) Warnings. The following sentences shall appear in boldface type as the first statement under the heading "Warnings."
(i) Oral dosage forms. "Taking more than the recommended dose in 24 hours can be harmful."
(ii) Rectal enema dosage forms. "Using more than one enema in 24 hours can be harmful."
(3) Directions - (i) The labeling of all orally or rectally administered OTC drug products containing sodium phosphates shall contain the following directions in boldface type immediately preceding the dosage information: "Do not" ("take" or "use") "more unless directed by a doctor. See Warnings."
(ii) For products containing dibasic sodium phosphate/monobasic sodium phosphate identified in § 334.16(d) marketed as a solution. Adults and children 12 years of age and over: Oral dosage is dibasic sodium phosphate 3.42 to 7.56 grams (g) and monobasic sodium phosphate 9.1 to 20.2 g (20 to 45 mL dibasic sodium phosphate/monobasic sodium phosphate oral solution) as a single daily dose. "Do not take more than 45 mL (9 teaspoonfuls or 3 tablespoonfuls) in a 24-hour period." Children 10 and 11 years of age: Oral dosage is dibasic sodium phosphate 1.71 to 3.78 g and monobasic sodium phosphate 4.5 to 10.1 g (10 to 20 mL dibasic sodium phosphate/monobasic sodium phosphate oral solution) as a single daily dose. "Do not take more than 20 mL (4 teaspoonfuls) in a 24-hour period." Children 5 to 9 years of age: Oral dosage is dibasic sodium phosphate 0.86 to 1.89 g and monobasic sodium phosphate 2.2 to 5.05 g (5 to 10 mL dibasic sodium phosphate/monobasic sodium phosphate oral solution) as a single daily dose. "Do not take more than 10 mL (2 teaspoonfuls) in a 24-hour period." Children under 5 years of age: ask a doctor.
(c) After June 22, 1998, for package size limitation and September 18, 1998, for labeling in accord with paragraph (b) of this section, any such OTC drug product initially introduced or initially delivered for introduction into interstate commerce, or any such drug product that is repackaged or relabeled after these dates regardless of the date the product was manufactured, initially introduced, or initially delivered for introduction into interstate commerce, that is not in compliance with this section is subject to regulatory action.
[63 FR 27843, May 21, 1998]
|
|
|
Sec. 201.308 Ipecac syrup; warnings and directions for use for over-the-counter sale.
|
|
(a) It is estimated that each year about 500,000 accidental poisonings occur in the United States and result in approximately 1,500 deaths, of which over 400 are children. In the emergency treatment of these poisonings, ipecac syrup is considered the emetic of choice. The immediate availability of this drug for use in such situations is critical, since rapid treatment may be the difference between life and death. The restriction of this drug to prescription sale limits its availability in emergencies. On the other hand, it is the consensus of informed medical opinion that ipecac syrup should be used only under medical supervision in the emergency treatment of poisonings. In view of these facts, the question of whether ipecac syrup labeled as an emergency treatment for use in poisonings should be available over the counter has been controversial.
(b) In connection with its study of this problem, the Food and Drug Administration has obtained the views of medical authorities. It is the unanimous recommendation of the American Academy of Pediatrics, the American Association of Poison Control Centers, the American Medical Association, and the Medical Advisory Board of the Food and Drug Administration that ipecac syrup in 1 fluid ounce containers be permitted to be sold without prescription so that it will be readily available in the household for emergency treatment of poisonings, under medical supervision, and that the drug be appropriately packaged and labeled for this purpose.
(c) In view of the above recommendations, the Commissioner of Food and Drugs has determined that it is in the interest of the public health for ipecac syrup to be available for sale without prescription, provided that it is packaged in a quantity of 1 fluid ounce (30 milliliters), and its label bears, in addition to other required label information, the following, in a prominent and conspicuous manner:
(1) A statement conspicuously boxed and in red letters, to the effect: "For emergency use to cause vomiting in poisoning. Before using, call physician, the Poison Control Center, or hospital emergency room immediately for advice."
(2) A warning to the effect: "Warning - Keep out of reach of children. Do not use in unconscious persons. Ordinarily, this drug should not be used if strychnine, corrosives such as alkalies (lye) and strong acids, or petroleum distillates such as kerosine, gasoline, coal oil, fuel oil, paint thinner, or cleaning fluid have been ingested."
(3) Usual dosage: 1 tablespoon (15 milliliters) in persons over 1 year of age.
|
|
|
Sec. 201.309 Acetophenetidin (phenacetin)-containing preparations; necessary warning statement.
|
|
(a) In 1961, the Food and Drug Administration, pursuant to its statutory responsibility for the safety and effectiveness of drugs shipped in interstate commerce, began an active investigation of reports of possible toxic effects and renal damage due to misuse of the drug acetophenetidin. This study led to the decision that there was probable cause to conclude that misuse and prolonged use of the drug were in fact responsible for kidney lesions and disease. The Commissioner of Food and Drugs, in December 1963, appointed an ad hoc Advisory Committee of Inquiry on Possible Nephrotoxicity Associated With the Abuse of Acetophenetidin (Phenacetin)-Containing Preparations. This committee, composed of scientists in the fields of pharmacology and medicine, on April 23, 1964, submitted its findings and conclusions in the matter and recommended that all acetophenetidin (phenacetin)-containing preparations bear a warning as provided in section 502(f)(2) of the Federal Food, Drug, and Cosmetic Act.
(b) On the basis of the studies made by the Food and Drug Administration and the report of the Advisory Committee, the Commissioner of Food and Drugs has concluded that it is necessary for the protection of users that the label and labeling of all acetophenetidin (phenacetin)-containing preparations bear a warning statement to the following effect: "Warning - This medication may damage the kidneys when used in large amounts or for a long period of time. Do not take more than the recommended dosage, nor take regularly for longer than 10 days without consulting your physician."
|
|
|
Sec. 201.310 Phenindione; labeling of drug preparations intended for use by man.
|
|
(a) Reports in the medical literature and data accumulated by the Food and Drug Administration indicate that phenindione, a synthetic anticoagulant drug, has caused a number of cases of agranulocytosis (with two fatalities). There are also reports implicating the drug in cases of hepatitis and hypersensitivity reactions. In view of the potentially serious effects found to be associated with preparations of this drug intended for use by man, the Commissioner of Food and Drugs will regard such preparations as misbranded within the meaning of section 502(f) (1) and (2) of the Federal Food, Drug, and Cosmetic Act, unless the label and labeling on or within the package from which the drug is to be dispensed, and any other labeling furnishing or purporting to furnish information for use of the drug, bear a conspicuous warning statement to the following effect: "Warning: Agranulocytosis and hepatitis have been associated with the use of phenindione. Patients should be instructed to report promptly prodromal symptoms such as marked fatigue, chill, fever, and sore throat. Periodic blood studies and liver function tests should be performed. Use of the drug should be discontinued if leukopenia occurs or if evidence of hypersensitivity, such as dermatitis or fever, appears."
(b) Regulatory action may be initiated with respect to preparations of phenindione intended for use by man found within the jurisdiction of the act on or after November 25, 1961, unless such preparations are labeled in accordance with paragraph (a) of this section.
|
|
|
|
|
Sec. 201.312 Magnesium sulfate heptahydrate; label declaration on drug products.
|
|
Magnesium sulfate heptahydrate should be listed on the label of a drug product as epsom salt, which is its common or usual name.
|
|
|
Sec. 201.313 Estradiol labeling.
|
|
The article presently recognized in The National Formulary under the heading "Estradiol" and which is said to be "17-cis-beta estradiol" is the same substance formerly recognized in the United States Pharmacopeia under the designation "Alpha Estradiol." The substance should no longer be referred to in drug labeling as "Alpha Estradiol." The Food and Drug Administration would not object to label references to the article as simply "Estradiol"; nor would it object if the label of a preparation containing this substance referred to the presence of "Estradiol (formerly known as Alpha Estradiol)."
|
|
|
Sec. 201.314 Labeling of drug preparations containing salicylates.
|
|
(a) The label of any oral drug preparation intended for sale without prescription and which contains any salicylate ingredient (including aspirin, salicylamide, other salicylates, and combinations) must conspicuously bear, on a clearly contrasting background, the warning statement: "Keep out of reach of children [highlighted in bold type]. In case of overdose, get medical help or contact a Poison Control Center right away," or "Keep out of reach of children [highlighted in bold type]," except that if the article is an aspirin preparation, it shall bear the first of these warning statements. Such a warning statement is required for compliance with section 502(f)(2) of the Federal Food, Drug, and Cosmetic Act and is intended to guard against accidental poisonings. Safety closures that prevent access to the drug by young children are also recommended to guard against accidental poisonings.
(b) Effervescent preparations and preparations containing para-aminosalicylate as the only salicylate ingredient are exempted from this labeling requirement.
(c) Aspirin tablets sold as such and containing no other active ingredients, except tablets which cannot be readily subdivided into a child's dose because of their coating or size, should always bear dosage directions for each age group down to 3 years of age, with a statement such as "For children under 3 years of age, consult your physician." It is recommended that:
(1) Aspirin tablets especially made for pediatric use be produced only in 1
1/4-grain size to reduce the hazard of errors in dosage;
(2) By June 1, 1967, manufacturers and distributors of 1
1/4-grain size aspirin tablets discontinue the distribution of such tablets in retail containers containing more than 36 tablets, to reduce the hazard of accidental poisoning;
(3) The flavoring of 5-grain aspirin tablets or other "adult aspirin tablets" be discontinued; and
(4) Labeling giving undue emphasis to the pleasant flavor of flavored aspirin tablets be discontinued.
(d) Salicylate preparations other than aspirin tablets sold as such may, at the option of the distributor, be labeled for use by adults only. If their labeling and advertising clearly offer them for administration to adults only.
(e)(1) It is the obligation of the distributor who labels a salicylate preparation for administration to children to make certain that the article is suitable for such use and labeled with adequate directions for use in the age group for which it is offered, but in no case should such an article bear directions for use in children under 3 years of age. If the directions provide for administration to children as young as 3 years of age, the label should bear the statement, "For children under 3 years of age consult your physician." However, if the directions provide for administration to children only of an age greater than 3 years (for example, the dosage instructions provide for administration of the article to children only down to age 6), the label should bear a statement such as, "For younger children consult your physician."
(2) A statement such as, "For children under 3 years of age consult your physician" or "For younger children consult your physician" is not required on the label of an article clearly offered for administration to adults only.
(f) If the labeling or advertising of a salicylate preparation offers it for use in arthritis or rheumatism, the label and labeling should clearly state that the beneficial effects claimed are limited to: "For the temporary relief of minor aches and pains of arthritis and rheumatism." The qualifying phrase "for the temporary relief of minor aches and pains" should appear with the same degree of prominence and conspicuousness as the phrase "arthritis and rheumatism". The label and labeling should bear in juxtaposition with such directions for use conspicuous warning statements to the effect: "Caution: If pain persists for more than 10 days, or redness is present, or in conditions affecting children under 12 years of age, consult a physician immediately." The salicylate dosage should not exceed 60 grains in a 24-hour period or 10 grains in a 4-hour period. If the article contains other analgesics, the salicylate dosage should be appropriately reduced.
(g)(1) The label of any drug containing more than 5 percent methyl salicylate (wintergreen oil) should bear a conspicuous warning such as: "Do not use otherwise than as directed." These drug products must also include the "Keep out of reach of children" warning and the accidental ingestion warning as required in § 330.1(g) of this chapter.
(2) If the preparation is a counterirritant or rubefacient, it should also bear a caution such as, "Caution: Discontinue use if excessive irritation of the skin develops. Avoid getting into the eyes or on mucous membranes." (See also § 201.303.)
(h)(1) The labeling of orally or rectally administered over-the-counter drug products containing aspirin or nonaspirin salicylates as active ingredients subject to this paragraph is required to prominently bear the following warning: "Reye's syndrome [subheading in bold type]: Children and teenagers who have or are recovering from chicken pox or flu-like symptoms should not use this product. When using this product, if changes in behavior with nausea and vomiting occur, consult a doctor because these symptoms could be an early sign of Reye's syndrome, a rare but serious illness."
(2) This warning statement shall appear on the immediate container labeling. In cases where the immediate container is not the retail package, the retail package also must bear the warning statement. In addition, the warning statement shall appear on any labeling that contains warnings and, in such cases, the warning statement shall be the first warning statement under the heading "Warnings."
(3) Over-the-counter drug products subject to this paragraph and labeled solely for use by children (pediatric products) shall not recommend the product for use in treating flu or chicken pox.
(4) Any product subject to paragraphs (h)(1), (h)(2), and (h)(3) of this section that is not labeled as required by these paragraphs and that is initially introduced or initially delivered for introduction into interstate commerce after the following dates is misbranded under sections 201(n) and 502(a) and (f) of the Federal Food, Drug, and Cosmetic Act.
(i) Compliance by October 18, 2004, for OTC drug products containing aspirin and nonaspirin salicylates as an active ingredient and marketed under a new drug application or abbreviated new drug application.
(ii) Compliance by April 19, 2004, for OTC antidiarrheal and overindulgence drug products that contain bismuth subsalicylate as an active ingredient and have annual sales greater than $25,000.
(iii) Compliance by April 18, 2005, for OTC antidiarrheal and overindulgence drug products that contain bismuth subsalicylate as an active ingredient and have annual sales less than $25,000.
(iv) Compliance dates for all other OTC drug products containing aspirin and nonaspirin salicylates as an active ingredient and marketed under an OTC drug monograph (for internal analgesic, antipyretic, and antirheumatic drug products, or for menstrual drug products) will be established when the final monographs for those products are published in a future issue of the Federal Register. In the interim, these products should continue to be labeled with the previous Reye's syndrome warning that appears in paragraph (h)(1) of this section.
[40 FR 13998, Mar. 27, 1985, as amended at 51 FR 8182, Mar. 7, 1986; 53 FR 21637, June 9, 1988; 53 FR 24830, June 30, 1988; 64 FR 13291, Mar. 17, 1999; 65 FR 8, Jan. 3, 2000; 68 FR 18869, Apr. 17, 2003]
|
|
|
Sec. 201.315 Over-the-counter drugs for minor sore throats; suggested warning.
|
|
The Food and Drug Administration has studied the problem of the labeling of lozenges or troches containing a local anesthetic, chewing gum containing aspirin, various mouth washes and gargles and other articles sold over the counter for the relief of minor irritations of the mouth or throat. It will not object to the labeling of suitable articles of this type "For the temporary relief of minor sore throats", provided this is immediately followed in the labeling with a warning statement in prominent type essentially as follows: "Warning - Severe or persistent sore throat or sore throat accompanied by high fever, headache, nausea, and vomiting may be serious. Consult physician promptly. Do not use more than 2 days or administer to children under 3 years of age unless directed by physician."
|
|
|
Sec. 201.316 Drugs with thyroid hormone activity for human use; required warning.
|
|
(a) Drugs with thyroid hormone activity have been promoted for, and continue to be dispensed and prescribed for, use in the treatment of obesity, although their safety and effectiveness for that use have never been established.
(b) Drugs for human use with thyroid hormone activity are misbranded within the meaning of section 502 of the Federal Food, Drug, and Cosmetic Act unless their labeling bears the following boxed warning at the beginning of the "Warnings" section:
Drugs with thyroid hormone activity, alone or together with other therapeutic agents, have been used for the treatment of obesity. In euthyroid patients, doses within the range of daily hormonal requirements are ineffective for weight reduction. Larger doses may produce serious or even life-threatening manifestations of toxicity, particularly when given in association with sympathomimetic amines such as those used for their anorectic effects. [43 FR 22009, May 23, 1978]
|
|
|
Sec. 201.317 Digitalis and related cardiotonic drugs for human use in oral dosage forms; required warning.
|
|
(a) Digitalis and related cardiotonic drugs for human use in oral dosage forms have been promoted for, and continue to be dispensed and prescribed for, use in the treatment of obesity, although their safety and effectiveness for that use have never been established.
(b) Digitalis and related cardiotonic drugs for human use in oral dosage forms are misbranded within the meaning of section 502 of the Federal Food, Drug, and Cosmetic Act unless their labeling bears the following boxed warning at the beginning of the "Warnings" section:
(c) This section does not apply to digoxin products for oral use.
Digitalis alone or with other drugs has been used in the treatment of obesity. This use of digoxin or other digitalis glycosides is unwarranted. Moreover, since they may cause potentially fatal arrhythmias or other adverse effects, the use of these drugs in the treatment of obesity is dangerous. [43 FR 22009, May 23, 1978, as amended at 85 FR 72907, Nov. 16, 2020]
|
|
|
Sec. 201.319 Water-soluble gums, hydrophilic gums, and hydrophilic mucilloids (including, but not limited to agar, alginic acid, calcium polycarbophil, carboxymethylcellulose sodium, carrageenan, chondrus, glucomannan ((B-1,4 linked) polymannose acetate), guar gum, karaya gum, kelp, methylcellulose, plantago seed (psyllium), polycarbophil tragacanth, and xanthan gum) as active ingredients; required warnings and directions.
|
|
(a) Reports in the medical literature and data accumulated by the Food and Drug Administration indicate that esophageal obstruction and asphyxiation have been associated with the ingestion of water-soluble gums, hydrophilic gums, and hydrophilic mucilloids including, but not limited to, agar, alginic acid, calcium polycarbophil, carboxymethylcellulose sodium, carrageenan, chondrus, glucomannan ((B-1,4 linked) polymannose acetate), guar gum, karaya gum, kelp, methylcellulose, plantago seed (psyllium), polycarbophil, tragacanth, and xanthan gum. Esophageal obstruction and asphyxiation due to orally-administered drug products containing water-soluble gums, hydrophilic gums, and hydrophilic mucilloids as active ingredients are significant health risks when these products are taken without adequate fluid or when they are used by individuals with esophageal narrowing or dysfunction, or with difficulty in swallowing. Additional labeling is needed for the safe and effective use of any OTC drug product for human use containing a water-soluble gum, hydrophilic gum, or hydrophilic mucilloid as an active ingredient when marketed in a dry or incompletely hydrated form to include, but not limited to, the following dosage forms: Capsules, granules, powders, tablets, and wafers. Granular dosage forms containing psyllium are not generally recognized as safe and effective as OTC laxatives (see § 310.545(a)(12)(i)(B) of this chapter) and may not be marketed without an approved new drug application because the warnings and directions in paragraph (b) of this section have been found inadequate for these products.
(b) Any drug products for human use containing a water-soluble gum, hydrophilic gum, or hydrophilic mucilloid as an active ingredient in an oral dosage form when marketed in a dry or incompletely hydrated form as described in paragraph (a) of this section are misbranded within the meaning of section 502 of the Federal Food, Drug, and Cosmetic Act unless their labeling bears the following warnings (under the subheading "Choking") and directions:
" 'Choking' [highlighted in bold type]: Taking this product without adequate fluid may cause it to swell and block your throat or esophagus and may cause choking. Do not take this product if you have difficulty in swallowing. If you experience chest pain, vomiting, or difficulty in swallowing or breathing after taking this product, seek immediate medical attention;" and
" 'Directions' [highlighted in bold type]:" (Select one of the following, as appropriate: "Take" or "Mix") "this product (child or adult dose) with at least 8 ounces (a full glass) of water or other fluid. Taking this product without enough liquid may cause choking. See choking warning."
(c) After February 28, 1994, any such OTC drug product initially introduced or initially delivered for introduction into interstate commerce, or any such drug product that is repackaged or relabeled after this date regardless of the date the product was manufactured, initially introduced, or initially delivered for introduction into interstate commerce, that is not in compliance with this section is subject to regulatory action.
[58 FR 45201, Aug. 26, 1993, as amended at 64 FR 13292, Mar. 17, 1999; 72 FR 14674, Mar. 29, 2007]
|
|
|
Sec. 201.320 Warning statements for drug products containing or manufactured with chlorofluorocarbons or other ozone-depleting substances.
|
|
(a)(1) All drug products containing or manufactured with chlorofluorocarbons, halons, carbon tetrachloride, methyl chloride, or any other class I substance designated by the Environmental Protection Agency (EPA) shall, except as provided in paragraph (b) or (c) of this section, bear the following warning statement:
Warning: Contains [or Manufactured with, if applicable] [insert name of substance ], a substance which harms public health and the environment by destroying ozone in the upper atmosphere.
(2) The warning statement shall be clearly legible and conspicuous on the product, its immediate container, its outer packaging, or other labeling in accordance with the requirements of 40 CFR part 82 and appear with such prominence and conspicuousness as to render it likely to be read and understood by consumers under normal conditions of purchase.
(b)(1) For prescription drug products for human use, the following alternative warning statement may be used:
(2) The warning statement shall be clearly legible and conspicuous on the product, its immediate container, its outer packaging, or other labeling in accordance with the requirements of 40 CFR part 82 and appear with such prominence and conspicuousness as to render it likely to be read and understood by consumers under normal conditions of purchase.
(3) If the warning statement in paragraph (b)(1) of this section is used, the following warning statement must be placed on the package labeling intended to be read by the physician (physician package insert) after the "How supplied" section, which describes special handling and storage conditions on the physician labeling:
(c)(1) For over-the-counter drug products for human use, the following alternative warning statement may be used:
(2) The warning statement shall be clearly legible and conspicuous on the product, its immediate container, its outer packaging, or other labeling in accordance with the requirements of 40 CFR part 82 and appear with such prominence and conspicuousness as to render it likely to be read and understood by consumers under normal conditions of purchase.
(d) This section does not replace or relieve a person from any requirements imposed under 40 CFR part 82.
The indented statement below is required by the Federal government's Clean Air Act for all products containing or manufactured with chlorofluorocarbons (CFC's) [or name of other class I substance, if applicable]:
This product contains [or is manufactured with, if applicable] [insert name of substance ], a substance which harms the environment by destroying ozone in the upper atmosphere.
Your physician has determined that this product is likely to help your personal health. USE THIS PRODUCT AS DIRECTED, UNLESS INSTRUCTED TO DO OTHERWISE BY YOUR PHYSICIAN. If you have any questions about alternatives, consult with your physician. Note: The indented statement below is required by the Federal government's Clean Air Act for all products containing or manufactured with chlorofluorocarbons (CFC's) [or name of other class I substance, if applicable]:
Warning: Contains [or Manufactured with, if applicable] [insert name of substance ], a substance which harms public health and the environment by destroying ozone in the upper atmosphere.
A notice similar to the above WARNING has been placed in the information for the patient [or patient information leaflet, if applicable] of this product under the Environmental Protection Agency's (EPA's) regulations. The patient's warning states that the patient should consult his or her physician if there are questions about alternatives. The indented statement below is required by the Federal government's Clean Air Act for all products containing or manufactured with chlorofluorocarbons (CFC's) [or other class I substance, if applicable]:
Warning: Contains [or Manufactured with, if applicable] [insert name of substance ], a substance which harms public health and environment by destroying ozone in the upper atmosphere.
CONSULT WITH YOUR PHYSICIAN OR HEALTH PROFESSIONAL IF YOU HAVE ANY QUESTION ABOUT THE USE OF THIS PRODUCT. [61 FR 20100, May 3, 1996]
|
|
|
Sec. 201.323 Aluminum in large and small volume parenterals used in total parenteral nutrition.
|
|
(a) The aluminum content of large volume parenteral (LVP) drug products used in total parenteral nutrition (TPN) therapy must not exceed 25 micrograms per liter ([micro]g/L).
(b) The package insert of LVP's used in TPN therapy must state that the drug product contains no more than 25 [micro]g/L of aluminum. This information must be contained in the "Precautions" section of the labeling of all large volume parenterals used in TPN therapy.
(c) Except as provided in paragraph (d) of this section, the maximum level of aluminum present at expiry must be stated on the immediate container label of all small volume parenteral (SVP) drug products and pharmacy bulk packages (PBPs) used in the preparation of TPN solutions. The aluminum content must be stated as follows: "Contains no more than __ [micro]g/L of aluminum." The immediate container label of all SVP's and PBP's that are lyophilized powders used in the preparation of TPN solutions must contain the following statement: "When reconstituted in accordance with the package insert instructions, the concentration of aluminum will be no more than __ [micro]g/L." This maximum level of aluminum must be stated as the highest of:
(1) The highest level for the batches produced during the last 3 years;
(2) The highest level for the latest five batches, or
(3) The maximum historical level, but only until completion of production of the first five batches after July 26, 2004.
(d) If the maximum level of aluminum is 25 [micro]g/L or less, instead of stating the exact amount of aluminum as required in paragraph (c) of this section, the immediate container label may state: "Contains no more than 25 [micro]g/L of aluminum." If the SVP or PBP is a lyophilized powder, the immediate container label may state: "When reconstituted in accordance with the package insert instructions, the concentration of aluminum will be no more than 25 [micro]g/L".
(e) The package insert for all LVP's, all SVP's, and PBP's used in TPN must contain a warning statement. This warning must be contained in the "Warnings" section of the labeling. The warning must state:
WARNING: This product contains aluminum that may be toxic. Aluminum may reach toxic levels with prolonged parenteral administration if kidney function is impaired. Premature neonates are particularly at risk because their kidneys are immature, and they require large amounts of calcium and phosphate solutions, which contain aluminum.
Research indicates that patients with impaired kidney function, including premature neonates, who receive parenteral levels of aluminum at greater than 4 to 5 [micro]g/kg/day accumulate aluminum at levels associated with central nervous system and bone toxicity. Tissue loading may occur at even lower rates of administration.
(f) Applicants and manufacturers must use validated assay methods to determine the aluminum content in parenteral drug products. The assay methods must comply with current good manufacturing practice requirements. Applicants must submit to the Food and Drug Administration validation of the method used and release data for several batches. Manufacturers of parenteral drug products not subject to an approved application must make assay methodology available to FDA during inspections. Holders of pending applications must submit an amendment under § 314.60 or § 314.96 of this chapter.
[65 FR 4110, Jan. 26, 2000, as amended at 67 FR 70691, Nov. 26, 2002; 68 FR 32981, June 3, 2003]
|
|
|
Sec. 201.325 Over-the-counter drugs for vaginal contraceptive and spermicide use containing nonoxynol 9 as the active ingredient; required warnings and labeling information.
|
|
(a) Studies indicate that use of vaginal contraceptive drug products containing nonoxynol 9 does not protect against infection from the human immunodeficiency virus (HIV), the virus that causes acquired immunodeficiency syndrome (AIDS), or against the transmission of other sexually transmitted diseases (STDs). Studies also indicate that use of vaginal contraceptive drug products containing nonoxynol 9 can increase vaginal irritation, such as the disruption of the vaginal epithelium, and also can cause epithelial disruption when used in the rectum. These effects may increase the risk of transmission of the AIDS virus (HIV) from an infected partner. Therefore, consumers should be warned that these products do not protect against the transmission of the AIDS virus (HIV) or other STDs, that use of these products can increase vaginal and rectal irritation, which may increase the risk of getting the AIDS virus (HIV) from an HIV infected partner, and that the products are not for rectal use. Consumers should also be warned that these products should not be used by persons who have HIV/AIDS or are at high risk for HIV/AIDS.
(b) The labeling of OTC vaginal contraceptive and spermicide drug products containing nonoxynol 9 as the active ingredient, whether subject to the ongoing OTC drug review or an approved drug application, must contain the following warnings under the heading "Warnings," in accordance with 21 CFR 201.66.
(1) "[bullet] For vaginal use only [bullet] Not for rectal (anal) use" [both warnings in bold type].
(2) "Sexually transmitted diseases (STDs) alert [in bold type]: This product does not [word "not" in bold type] protect against HIV/AIDS or other STDs and may increase the risk of getting HIV from an infected partner".
(3) "Do not use" [in bold type] if you or your sex partner has HIV/AIDS. If you do not know if you or your sex partner is infected, choose another form of birth control".
(4) "When using this product [in bold type] [optional, bullet] you may get vaginal irritation (burning, itching, or a rash)".
(5) "Stop use and ask a doctor if [in bold type] [optional, bullet] you or your partner get burning, itching, a rash, or other irritation of the vagina or penis".
(c) The labeling of this product states under the "Other information" section of the Drug Facts labeling in accordance with § 201.66(c)(7), "[bullet] when used correctly every time you have sex, latex condoms greatly reduce, but do not eliminate, the risk of catching or spreading HIV, the virus that causes AIDS.
(d) The labeling of this product includes the following statements either on the outside container or wrapper of the retail package, under the "Other information" section of the Drug Facts labeling in accordance with § 201.66(c)(7), or in a package insert:
(1) "[bullet] studies have raised safety concerns that products containing the spermicide nonoxynol 9 can irritate the vagina and rectum. Sometimes this irritation has no symptoms. This irritation may increase the risk of getting HIV/AIDS from an infected partner".
(2) "[bullet] you can use nonoxynol 9 for birth control with or without a diaphragm or condom if you have sex with only one partner who is not infected with HIV and who has no other sexual partners or HIV risk factors".
(3) "[bullet] use a latex condom without nonoxynol 9 if you or your sex partner has HIV/AIDS, multiple sex partners, or other HIV risk factors".
(4) "[bullet] ask a health professional if you have questions about your best birth control and STD prevention methods".
(e) Any drug product subject to this section that is not labeled as required and that is initially introduced or initially delivered for introduction into interstate commerce after June 19, 2008, is misbranded under section 502 of the Federal Food, Drug, and Cosmetic Act (the act) (21 U.S.C. 352), is a new drug under section 505 of the act (21 U.S.C. 355), and is subject to regulatory action.
[72 FR 71785, Dec. 19, 2007]
|
|
|
Sec. 201.326 Over-the-counter drug products containing internal analgesic/antipyretic active ingredients; required warnings and other labeling.
|
|
(a) Labeling. The labeling for all over-the-counter (OTC) drug products containing any internal analgesic/antipyretic active ingredients (including, but not limited to, acetaminophen, aspirin, carbaspirin calcium, choline salicylate, ibuprofen, ketoprofen, magnesium salicylate, naproxen sodium, and sodium salicylate) alone or in combination must bear the following labeling in accordance with §§ 201.60, 201.61, and 201.66.
(1) Acetaminophen - (i) Statement of identity. The statement of identity appears in accord with §§ 201.61 and 299.4 of this chapter. The ingredient name "acetaminophen" must appear highlighted (e.g., fluorescent or color contrast) or in bold type, be in lines generally parallel to the base on which the package rests as it is designed to be displayed, and be in one of the following sizes, whichever is greater:
(A) At least one-quarter as large as the size of the most prominent printed matter on the principal display panel (PDP), or
(B) At least as large as the size of the "Drug Facts" title, as required in § 201.66(d)(2). The presence of acetaminophen must appear as part of the established name of the drug, as defined in § 299.4 of this chapter. Combination products containing acetaminophen and a nonanalgesic ingredient(s) (e.g., cough-cold) must include the name "acetaminophen" and the name(s) of the other active ingredient(s) in the product on the PDP in accord with this paragraph. Only the name "acetaminophen" must appear highlighted or in bold type, and in a prominent print size, as described in this paragraph.
(ii) Active Ingredient and Purpose Headings. The information required under § 201.66(c)(2) and (c)(3) of this chapter must be included under these headings. The information under these headings, but not the headings, may appear highlighted.
(iii) For products labeled for adults only. The labeling of the product states the following warnings under the heading "Warnings":
(A) The liver warning states "Liver warning [heading in bold type]: This product contains acetaminophen. Severe liver damage may occur if you take [bullet] more than [insert maximum number of daily dosage units] in 24 hours, which is the maximum daily amount [optional: 'for this product'] [bullet] with other drugs containing acetaminophen [bullet] 3 or more alcoholic drinks every day while using this product". This "Liver" warning must be the first warning under the "Warnings" heading. For products that contain both acetaminophen and aspirin, this "Liver" warning must appear after the "Reye's syndrome" and "Allergy alert" warnings in § 201.66(c)(5)(ii)(A) and (c)(5)(ii)(B) and before the "Stomach bleeding" warning in paragraph (a)(2)(iii)(A) of this section. If there is an outer and immediate container of a retail package, this warning must appear on both the outer and immediate containers. If the immediate container is a blister card, the warning must appear on the blister card and remain intact and readable when drug product is removed from the blister card. The warning does not need to be included on each blister unit.
(B) "Do not use with any other drug containing acetaminophen (prescription or nonprescription). If you are not sure whether a drug contains acetaminophen, ask a doctor or pharmacist."
(C) "Ask a doctor before use if you have liver disease".
(D) "Ask a doctor or pharmacist before use if you are taking the blood thinning drug warfarin" except on the labeling of combination products that contain acetaminophen and NSAID(s).
(iv) For products labeled only for children under 12 years of age.
(A) Warnings. The labeling of the product states the following warnings under the heading "Warnings":
(1 ) The liver warning states "Liver warning [heading in bold type]: This product contains acetaminophen. Severe liver damage may occur if your child takes [bullet] more than 5 doses in 24 hours, which is the maximum daily amount [optional: 'for this product'] [bullet] with other drugs containing acetaminophen". This "Liver" warning must be the first warning under the "Warnings" heading. If there is an outer and immediate container of a retail package, this warning must appear on both the outer and immediate containers. If the immediate container is a blister card, the warning must appear on the blister card and remain intact and readable when drug product is removed from the blister card. The warning is not required to be included on each blister unit.
(2 ) "Do not use with any other drug containing acetaminophen (prescription or nonprescription). If you are not sure whether a drug contains acetaminophen, ask a doctor or pharmacist."
(3 ) "Ask a doctor before use if your child has liver disease".
(4 ) "Ask a doctor or pharmacist before use if your child is taking the blood thinning drug warfarin" except on the labeling of combination products that contain acetaminophen and NSAID(s).
(B) Directions. The labeling of the product contains the following information under the heading "Directions": "this product does not contain directions or complete warnings for adult use" [in bold type].
(v) For products labeled for adults and children under 12 years of age. The labeling of the product states all of the warnings in paragraphs (a)(1)(iii)(A), (a)(1)(iii)(B), and (a)(1)(iii)(C) of this section with the following modifications:
(A) The liver warning states "Liver warning [heading in bold type]: This product contains acetaminophen. Severe liver damage may occur if [bullet] adult takes more than [insert maximum number of daily dosage units] in 24 hours, which is the maximum daily amount [optional: 'for this product'] [bullet] child takes more than 5 doses in 24 hours [bullet] taken with other drugs containing acetaminophen [bullet] adult has 3 or more alcoholic drinks everyday while using this product." If there is an outer and immediate container of a retail package, this warning must appear on both the outer and immediate containers. If the immediate container is a blister card, the warning must appear on the blister card and remain intact and readable when drug product is removed from the blister card. The warning is not required to be included on each blister unit.
(B) "Ask a doctor before use if the user has liver disease."
(C) "Do not use with any other drug containing acetaminophen (prescription or nonprescription). If you are not sure whether a drug contains acetaminophen, ask a doctor or pharmacist."
(D) "Ask a doctor or pharmacist before use if the user is taking the blood thinning drug warfarin" except on the labeling of combination products that contain acetaminophen and NSAID(s).
(2) Nonsteroidal anti-inflammatory analgesic/antipyretic active ingredients - including, but not limited to, aspirin, carbaspirin calcium, choline salicylate, ibuprofen, ketoprofen, magnesium salicylate, naproxen sodium, and sodium salicylate.
(i) Statement of identity. The statement of identity appears in accord with §§ 201.61 and 299.4 of this chapter. The word "(NSAID)" must appear highlighted (e.g., fluorescent or color contrast) or in bold type, be in lines generally parallel to the base on which the package rests as it is designed to be displayed, and be in one of the following sizes, whichever is greater:
(A) At least one-quarter as large as the size of the most prominent printed matter on the PDP, or
(B) At least as large as the size of the "Drug Facts" title, as required in § 201.66(d)(2). The word "(NSAID)" must appear as part of the established name of the drug, as defined in § 299.4 of this chapter, or after the general pharmacological (principal intended) action of the NSAID ingredient. Combination products containing an NSAID and a nonanalgesic ingredient(s) (e.g., cough-cold) must include the name of the NSAID ingredient and the word "(NSAID)" in accordance with this paragraph, and the name(s) of the other active ingredient(s) in the product on the PDP. Only the word "(NSAID)" needs to appear highlighted or in bold type, and in a prominent print size, as described in this paragraph.
(ii) Active Ingredient and Purpose Headings. The information required under § 201.66(c)(2) and (c)(3) of this chapter must be included under these headings. The active ingredient(s) section of the product's labeling, as defined in § 201.66(c)(2), contains the term "(NSAID*)" after the NSAID active ingredient with an asterisk statement at the end of the active ingredient(s) section that defines the term "NSAID" and states "* nonsteroidal anti-inflammatory drug." The information under these headings may appear highlighted. However, the headings "Active Ingredient" and "Purpose" may not appear highlighted.
(iii) For products labeled for adults only. The labeling of the product states the following warnings under the heading "Warnings":
(A) The stomach bleeding warning states "Stomach bleeding warning [heading in bold type]: This product contains an NSAID, which may cause severe stomach bleeding. The chance is higher if you [bullet] are age 60 or older [bullet] have had stomach ulcers or bleeding problems [bullet] take a blood thinning (anticoagulant) or steroid drug [bullet] take other drugs containing prescription or nonprescription NSAIDs (aspirin, ibuprofen, naproxen, or others) [bullet] have 3 or more alcoholic drinks every day while using this product [bullet] take more or for a longer time than directed". This "Stomach bleeding" warning must appear after the "Reye's syndrome" and "Allergy alert" warnings in § 201.66(c)(5)(ii)(A) and (c)(5)(ii)(B). For products that contain both acetaminophen and aspirin, the acetaminophen "Liver" warning in paragraph (a)(1)(iii) of this section must appear before the "Stomach bleeding" warning in this paragraph. If there is an outer and immediate container of a retail package, this warning must appear on both the outer and immediate containers. If the immediate container is a blister card, the warning must appear on the blister card and remain intact and readable when drug product is removed from the blister card. The warning is not required to be included on each blister unit.
(B) "Ask a doctor before use if [bullet] stomach bleeding warning applies to you [bullet] you have a history of stomach problems, such as heartburn [bullet] you have high blood pressure, heart disease, liver cirrhosis, or kidney disease [bullet] you are taking a diuretic".
(C) "Stop use and ask a doctor if [bullet] you experience any of the following signs of stomach bleeding:" [add the following as second level of statements: "[bullet] feel faint [bullet] vomit blood [bullet] have bloody or black stools [bullet] have stomach pain that does not get better"].
(iv) For products labeled only for children under 12 years of age.
(A) Warnings. The labeling of the product states the following warnings under the heading "Warnings":
(1 ) The stomach bleeding warning states "Stomach bleeding warning [heading in bold type]: This product contains an NSAID, which may cause severe stomach bleeding. The chance is higher if your child [bullet] has had stomach ulcers or bleeding problems [bullet] takes a blood thinning (anticoagulant) or steroid drug [bullet] takes other drugs containing prescription or nonprescription NSAIDs (aspirin, ibuprofen, naproxen, or others) [bullet] takes more or for a longer time than directed". The "Stomach bleeding" warning must appear after the "Reye's syndrome" and "Allergy alert" warnings in § 201.66(c)(5)(ii)(A) and (c)(5)(ii)(B). If there is an outer and immediate container of a retail package, this warning must appear on both the outer and immediate containers. If the immediate container is a blister card, the warning must appear on the blister card and remain intact and readable when drug product is removed from the blister card. The warning is not required to be included on each blister unit.
(2 ) "Ask a doctor before use if [bullet] stomach bleeding warning applies to your child [bullet] child has a history of stomach problems, such as heartburn [bullet] child has not been drinking fluids [bullet] child has lost a lot of fluid due to vomiting or diarrhea [bullet] child has high blood pressure, heart disease, liver cirrhosis, or kidney disease [bullet] child is taking a diuretic".
(3 ) "Stop use and ask a doctor if [bullet] child experiences any of the following signs of stomach bleeding:" [add the following as second level of statements: [bullet] feels faint [bullet] vomits blood [bullet] has bloody or black stools [bullet] has stomach pain that does not get better"].
(B) Directions. The labeling of the product contains the following information under the heading "Directions": "this product does not contain directions or complete warnings for adult use" [in bold type].
(v) For products labeled for adults and children under 12 years of age. The labeling of the product states all of the warnings in paragraphs (a)(2)(iii)(A) through (a)(2)(iii)(C) of this section with the following modifications:
(A) The Stomach bleeding warning states "Stomach bleeding warning [heading in bold type]: This product contains an NSAID, which may cause severe stomach bleeding. The chance is higher if the user [bullet] has had stomach ulcers or bleeding problems [bullet] takes a blood thinning (anticoagulant) or steroid drug [bullet] takes other drugs containing prescription or nonprescription NSAIDs (aspirin, ibuprofen, naproxen, or others) [bullet] takes more or for a longer time than directed [bullet] is age 60 or older [bullet] has 3 or more alcoholic drinks everyday while using this product". The "Stomach bleeding" warning must appear after the "Reye's syndrome" and "Allergy alert" warnings in § 201.66(c)(5)(ii)(A) and (c)(5)(ii)(B). If there is an outer and immediate container of a retail package, this warning must appear on both the outer and immediate containers. If the immediate container is a blister card, the warning must appear on the blister card and remain intact and readable when drug product is removed from the blister card. The warning is not required to be included on each blister unit.
(B) The labeling states "Ask a doctor before use if [bullet] stomach bleeding warning applies to user [bullet] user has history of stomach problems, such as heartburn [bullet] user has high blood pressure, heart disease, liver cirrhosis, or kidney disease [bullet] user takes a diuretic [bullet] user has not been drinking fluids [bullet] user has lost a lot of fluid due to vomiting or diarrhea".
(C) The labeling states "Stop use and ask a doctor if [bullet] user experiences any of the following signs of stomach bleeding:" [add the following as second level of statements: [bullet] feels faint [bullet] vomits blood [bullet] has bloody or black stools [bullet] has stomach pain that does not get better"].
(b) New warnings information statement. The labeling of any drug product subject to this section that is initially introduced or initially delivered for introduction into interstate commerce before or on April 29, 2010, must bear on its PDP, as defined in § 201.60, the statement "See new warnings information". This statement must appear highlighted (e.g., fluorescent or color contrast) or in bold type, be in lines generally parallel to the base on which the package rests as it is designed to be displayed, and be in one of the following sizes, whichever is greater:
(1) At least one-quarter as large as the size of the most prominent printed matter on the PDP, or
(2) At least as large as the size of the "Drug Facts" title, as required in § 201.66(d)(2). The new warnings information statement must remain on the PDP of the drug product for at least 1 year from the date the product is initially introduced into interstate commerce.
(c) Requirements to supplement approved application. Holders of approved applications for OTC drug products that contain internal analgesic/antipyretic active ingredients that are subject to the requirements of paragraph (a) of this section must submit supplements under § 314.70(c) of this chapter to include the required information in the product's labeling. Such labeling may be put into use without advance approval of FDA provided it includes at least the exact information included in paragraph (a) of this section.
[74 FR 19407, Apr. 29, 2009, as amended at 74 FR 31180, June 30, 2009; 74 FR 61514, Nov. 25, 2009]
|
|
|
Sec. 201.327 Over-the-counter sunscreen drug products; required labeling based on effectiveness testing.
|
|
The following provisions apply to sunscreen products containing aminobenzoic acid, avobenzone, cinoxate, dioxybenzone, ensulizole, homosalate, meradimate, octinoxate, octisalate, octocrylene, oxybenzone, padimate O, sulisobenzone, titanium dioxide, trolamine salicylate, or zinc oxide, alone or in combination. The provisions do not apply to sunscreen products marketed under approved new drug applications or abbreviated new drug applications.
(a) Principal display panel. In addition to the statement of identity in paragraph (b) of this section, the following labeling shall be prominently placed on the principal display panel:
(1) Effectiveness claim - (i) For products that pass the broad spectrum test in paragraph (j) of this section. (A) The labeling states "Broad Spectrum SPF [insert numerical SPF value resulting from testing under paragraph (i) of this section]".
(B) Prominence. The Broad Spectrum SPF statement shall appear as continuous text with no intervening text or graphic. The entire text shall appear in the same font style, size, and color with the same background color.
(ii) For sunscreen products that do not pass the broad spectrum test in paragraph (j) of this section. The labeling states "SPF [insert numerical SPF value resulting from testing under paragraph (i) of this section]". The entire text shall appear in the same font style, size, and color with the same background color.
(2) Water resistance statements - (i) For products that provide 40 minutes of water resistance according to the test in paragraph (i)(7)(i) of this section. The labeling states "Water Resistant (40 minutes)".
(ii) For products that provide 80 minutes of water resistance according to the test in paragraph (i)(7)(ii) of this section. The labeling states "Water Resistant (80 minutes)".
(b) Statement of identity. The labeling of the product contains the established name of the drug, if any, and identifies the drug as a "sunscreen."
(c) Indications. The labeling of the product states, under the heading "Uses," the phrases listed in this paragraph (c), as appropriate. Other truthful and nonmisleading statements, describing only the uses that have been established and listed in this paragraph (c), may also be used, as provided in § 330.1(c)(2) of this chapter, subject to the provisions of section 502 of the Federal Food, Drug, and Cosmetic Act (the FD&C Act) relating to misbranding and the prohibition in section 301(d) of the FD&C Act against the introduction or delivery for introduction into interstate commerce of unapproved new drugs in violation of section 505(a) of the FD&C Act.
(1) For all sunscreen products, the following indication statement must be included under the heading "Uses": "[Bullet] helps prevent sunburn". See § 201.66(b)(4) of this chapter for definition of bullet.
(2) For sunscreen products with a Broad Spectrum SPF value of 15 or higher according to the tests in paragraphs (i) and (j) of this section, the labeling may include the following statement in addition to the indication in § 201.327(c)(1): "[Bullet] if used as directed with other sun protection measures (see Directions [in bold italic font]), decreases the risk of skin cancer and early skin aging caused by the sun".
(3) Any labeling or promotional materials that suggest or imply that the use, alone, of any sunscreen reduces the risk of or prevents skin cancer or early skin aging will cause the product to be misbranded under section 502 of the FD&C Act (21 U.S.C. 352).
(d) Warnings. The labeling of the product contains the following warnings under the heading "Warnings".
(1) For all sunscreen products. (i) The labeling states "Do not use [bullet] on damaged or broken skin".
(ii) The labeling states "When using this product [bullet] keep out of eyes. Rinse with water to remove."
(iii) The labeling states "Stop use and ask a doctor if [bullet] rash occurs".
(2) For sunscreen products that are broad spectrum with SPF values of at least 2 but less than 15 according to the SPF test in paragraph (i) of this section or that do not pass the broad spectrum test in paragraph (j) of this section. The first statement under the heading "Warnings" states "Skin Cancer/Skin Aging Alert [in bold font]; Spending time in the sun increases your risk of skin cancer and early skin aging. This product has been shown only to help prevent sunburn, not [in bold font] skin cancer or early skin aging."
(e) Directions. The labeling of the product contains the following statements, as appropriate, under the heading "Directions." More detailed directions applicable to a particular product formulation may also be included.
(1) For all sunscreen products. (i) As an option, the labeling may state "For sunscreen use:".
(ii) The labeling states "[bullet] apply [select one of the following: 'Liberally' or 'generously'] [and, as an option: 'And evenly'] 15 minutes before sun exposure".
(iii) As an option, the labeling may state "[bullet] apply to all skin exposed to the sun".
(iv) The labeling states "[bullet] children under 6 months of age: Ask a doctor".
(2) For sunscreen products with a Broad Spectrum SPF value of 15 or higher according to the tests in paragraphs (i) and (j) of this section. The labeling states "[bullet] Sun Protection Measures. [in bold font] Spending time in the sun increases your risk of skin cancer and early skin aging. To decrease this risk, regularly use a sunscreen with a Broad Spectrum SPF value of 15 or higher and other sun protection measures including: [Bullet] limit time in the sun, especially from 10 a.m.-2 p.m. [bullet] wear long-sleeved shirts, pants, hats, and sunglasses".
(3) For products that satisfy the water resistance test in paragraph (i)(7) of this section. The labeling states "[bullet] reapply: [Bullet] after [select one of the following determined by water resistance test: '40 minutes of' or '80 minutes of'] swimming or sweating [bullet] immediately after towel drying [bullet] at least every 2 hours".
(4) For products that do not satisfy the water resistance test in paragraph (i)(7) of this section. The labeling states "[bullet] reapply at least every 2 hours [bullet] use a water resistant sunscreen if swimming or sweating".
(f) Other information. The labeling of the product contains the following statement under the heading "Other information:" "[bullet] protect the product in this container from excessive heat and direct sun".
(g) False and misleading claims. There are claims that would be false and/or misleading on sunscreen products. These claims include but are not limited to the following: "Sunblock," "sweatproof," and "waterproof." These or similar claims will cause the product to be misbranded under section 502 of the FD&C Act (21 U.S.C. 352).
(h) Labeling of products containing a combination of sunscreen and skin protectant active ingredients. Statements of identity, indications, warnings, and directions for use, respectively, applicable to each ingredient in the product may be combined to eliminate duplicative words or phrases so that the resulting information is clear and understandable. Labeling provisions in § 347.50(e) of this chapter shall not apply to these products.
(i) SPF test procedure - (1) UV source (solar simulator). (i) Emission spectrum. A single port or multiport solar simulator should be filtered so that it provides a continuous emission spectrum from 290 to 400 nanometers (nm) with a limit of 1,500 Watts per square meter (W/m
2) on total irradiance for all wavelengths between 250 and 1,400 nm.
(A) The solar simulator should have the following percentage of erythema-effective radiation in each specified range of wavelengths:
Solar Simulator Emission Spectrum
| Wavelength range (nm)
| Percent erythemal contribution
1
|
|---|
| <290 | <0.1
| | 290-300 | 1.0-8.0
| | 290-310 | 49.0-65.0
| | 290-320 | 85.0-90.0
| | 290-330 | 91.5-95.5
| | 290-340 | 94.0-97.0
| | 290-400 | 99.9-100.0
|
(B) In addition, UVA II (320-340 nm) irradiance should equal or exceed 20 percent of the total UV (290-400 nm) irradiance. UVA I (340-400 nm) irradiance should equal or exceed 60 percent of the total UV irradiance.
(ii) Erythema action spectrum. (A) Calculate the erythema action spectrum weighting factor (Vi) at each wavelength λ:
(1 ) Vi (λ) = 1.0 (250 <λ �298 nm)
(2 ) Vi (λ) = 10
0.094* (
298â??λ) (298 <λ â?¤328 nm)
(3 ) Vi (λ) = 10
0.015* (
140â??λ) (328 <λ â?¤400 nm)
(B) Calculate the erythema-effective UV dose (E) delivered by a solar simulator as follows:
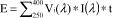
Where Vi(λ) = erythema action spectrum weighting factor at each wavelength λ
I(λ) = irradiance (Watts per square meter) at each wavelength λ
t = exposure time (seconds)
Erythema-effective dose (E) is expressed as effective Joules per square meter (J/m
2-eff).
(C) The emission spectrum must be determined using a handheld radiometer with a response weighted to match the spectrum in ISO 17166 CIE S 007/E entitled "Erythemal reference action spectrum and standard erythema dose," dated 1999 (First edition, 1999-12-15; corrected and reprinted 2000-11-15), which is incorporated by reference in accordance with 5 U.S.C. 552(a) and 1 CFR part 51. You may obtain a copy from the ISO Copyright Office, Case Postale 56, CH-1211, Geneva 20, Switzerland, telephone +41-22-749-01-11 or fax +41-22-74-09-47. http://www.iso.org. You may inspect a copy at the Center for Drug Evaluation and Research, 10903 New Hampshire Ave., Bldg. 22, Silver Spring, MD 20993, call 301-796-2090, or at the National Archives and Records Administration (NARA). For information on the availability of this material at NARA, call 202-741-6030, or go to: http://www.archives.gov/federal_register/code_of_federal_regulations/ibr_locations.html. The solar simulator output should be measured before and after each phototest or, at a minimum, at the beginning and end of each test day. This radiometer should be calibrated using side-by-side comparison with the spectroradiometer (using the weighting factors determined according to paragraph (i)(1)(ii)(A) of this section) at the time of the annual spectroradiometric measurement of the solar simulator as described in paragraph (i)(1)(iv) of this section.
(iii) Operation. A solar simulator should have no significant time-related fluctuations (within 20 percent) in radiation emissions after an appropriate warm-up time and demonstrate good beam uniformity (within 20 percent) in the exposure plane. The delivered dose to the UV exposure site must be within 10 percent of the expected dose.
(iv) Periodic measurement. To ensure that the solar simulator delivers the appropriate spectrum of UV radiation, the emission spectrum of the solar simulator should be measured at least annually with an appropriate and accurately calibrated spectroradiometer system (results should be traceable to the National Institute for Standards and Technology). In addition, the solar simulator must be recalibrated if there is any change in the lamp bulb or the optical filtering components (i.e., filters, mirrors, lenses, collimating devices, or focusing devices). Daily solar simulator radiation intensity should be monitored with a broadband radiometer with a response weighted to match the erythema action spectrum in ISO 17166 CIE S 007/E entitled "Erythemal reference action spectrum and standard erythema dose," which is incorporated by reference in paragraph (i)(1)(ii)(C) of this section. If a lamp must be replaced due to failure or aging during a phototest, broadband device readings consistent with those obtained for the original calibrated lamp will suffice until measurements can be performed with the spectroradiometer at the earliest possible opportunity.
(2) SPF standard - (i) Preparation. The SPF standard should be a formulation containing 7-percent padimate O and 3-percent oxybenzone.
Composition of the Padimate O/oxybenzone SPF Standard
| Ingredients
| Percent by weight
|
|---|
| Part A:
| | | Lanolin | 4.50
| | Cocoa butter | 2.00
| | Glyceryl monostearate | 3.00
| | Stearic acid | 2.00
| | Padimate O | 7.00
| | Oxybenzone | 3.00
| | Part B:
| | | Purified water USP | 71.60
| | Sorbitol solution | 5.00
| | Triethanolamine, 99 percent | 1.00
| | Methylparaben | 0.30
| | Propylparaben | 0.10
| | Part C:
| | | Benzyl alcohol | 0.50
| | Part D:
| | | Purified water USP | QS
1
|
Step 1. Add the ingredients of Part A into a suitable stainless steel kettle equipped with a propeller agitator. Mix at 77 to 82 deg.C until uniform.
Step 2. Add the water of Part B into a suitable stainless steel kettle equipped with a propeller agitator and begin mixing at 77 to 82 deg.C. Add the remaining ingredients of Part B and mix until uniform.
Step 3. Add the batch of Step 1 to the batch of Step 2 and mix at 77 to 82 deg.C until smooth and uniform. Slowly cool the batch to 49 to 54 deg.C.
Step 4. Add the benzyl alcohol of Part C to the batch of Step 3 at 49 to 54 deg.C. Mix until uniform. Continue to cool batch to 35 to 41 deg.C.
Step 5. Add sufficient water of Part D to the batch of Step 4 at 35 to 41 deg.C to obtain 100 grams of SPF standard. Mix until uniform. Cool batch to 27 to 32 deg.C.
(ii) HPLC assay. Use the following high performance liquid chromatography (HPLC) procedure to verify the concentrations of padimate O and oxybenzone in the SPF standard:
(A) Instrumentation. (1 ) Equilibrate a suitable liquid chromatograph to the following or equivalent conditions:
| (i) Column | C-18, 250 millimeters (mm) length, 4.6 mm inner diameter (5 microns)
| | (ii) Mobile Phase | 85:15:0.5 methanol: water: acetic acid
| | (iii) Flow Rate | 1.5 milliliters (mL) per minute
| | (iv) Temperature | Ambient
| | (v) Detector | UV spectrophotometer at 308 nanometers
| | (vi) Attenuation | As needed |
(2 ) Use HPLC grade reagents for mobile phase.
(B) Preparation of the HPLC reference standard. (1 ) Weigh 0.50 gram (g) of oxybenzone USP reference standard into a 250-mL volumetric flask. Dissolve and dilute to volume with isopropanol. Mix well.
(2 ) Weigh 0.50 g of padimate O USP reference standard into a 250-mL volumetric flask. Dissolve and dilute to volume with isopropanol. Mix well.
(3 ) Pipet 3.0 mL of the oxybenzone solution and 7.0 mL of the padimate O solution into a 100-mL volumetric flask. Dilute to volume with isopropanol and mix well.
(C) HPLC system suitability. (1 ) Make three replicate 10-microliter injections of the HPLC reference standard (described in paragraph (i)(2)(ii)(B) of this section). The relative standard deviation in peak areas should not be more than 2.0 percent for either oxybenzone or padimate O.
(2 ) Calculate the resolution (R) between the oxybenzone and padimate O peaks from one chromatogram as follows:
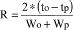
Where to = retention time for oxybenzone
tp = retention time for padimate O
Wo = oxybenzone peak width at baseline
Wp = padimate O peak width at baseline
If the resolution (R) is less than 3.0, adjust the mobile phase or replace the column.
(D) SPF standard assay - (1 ) The SPF standard is diluted to the same concentration as the HPLC reference standard according to the following steps:
(i ) Step 1. Weigh 1.0 g of the SPF standard (described in paragraph (i)(2)(i) of this section) into a 50-mL volumetric flask.
(ii ) Step 2. Add approximately 30 mL of isopropanol and heat with swirling until contents are evenly dispersed.
(iii ) Step 3. Cool to room temperature (15 to 30 deg.C) and dilute to volume with isopropanol. Mix well.
(iv ) Step 4. Pipet 5.0 mL of the preparation into a 50-mL volumetric flask and dilute to volume with isopropanol. Mix well.
(2 )(i ) Inject 10-microliter of diluted SPF standard from paragraph (i)(2)(D)(1 ) of this section and calculate the amount of oxybenzone and padimate O as follows:

(ii ) The percent of oxybenzone and padimate O in the SPF standard should be between 95 and 105.
(3) Test subjects - (i) Number of subjects. A test panel should include enough subjects to produce a minimum of 10 valid test results. A maximum of three subjects may be rejected from this panel based on paragraph (i)(5)(v)) of this section.
(ii) Medical history. (A) Obtain a medical history from each subject with emphasis on the effects of sunlight on the subject's skin. Determine that each subject is in good general health with skin type I, II, or III as follows:
(1 ) Always burns easily; never tans (sensitive).
(2 ) Always burns easily; tans minimally (sensitive).
(3 ) Burns moderately; tans gradually (light brown) (normal).
(4 ) Burns minimally; always tans well (moderate brown) (normal).
(5 ) Rarely burns; tans profusely (dark brown) (insensitive).
(6 ) Never burns; deeply pigmented (insensitive).
(B) Skin type is based on first 30 to 45 minutes of sun exposure after a winter season of no sun exposure. Determine that each subject is not taking topical or systemic medication that is known to alter responses to UV radiation. Determine that each subject has no history of sensitivities to topical products and/or abnormal responses to sunlight, such as a phototoxic or photoallergic response.
(iii) Physical examination. Conduct a physical examination to determine the presence of sunburn, suntan, scars, active dermal lesions, and uneven skin tones on the areas of the back to be tested. A suitable source of low power UVA, such as a Woods lamp, is helpful in this process. If any of these conditions are present, the subject is not qualified to participate in the study. The presence of nevi, blemishes, or moles will be acceptable if, in the physician's judgment, they will neither compromise the study nor jeopardize a subject's safety. Subjects with dysplastic nevi should not be enrolled. Excess hair on the back is acceptable if the hair is clipped. Shaving is unacceptable because it may remove a significant portion of the stratum corneum and temporarily alter the skin's response to UV radiation.
(iv) Informed consent. Obtain legally effective written informed consent from all test subjects.
(4) Sunscreen application. (i) Test site. Test sites are locations on each subject's back, between the beltline and the shoulder blades (scapulae) and lateral to the midline, where skin responses to UV radiation are determined. Responses on unprotected skin (no test material applied) and protected skin (sunscreen test product(s) or SPF standard applied) are determined at separate unprotected and protected test sites, respectively. Test sites should be randomly located in a blinded manner. Each test site should be a minimum of 30 square centimeters and outlined with indelible ink.
(ii) Test subsite. Test subsites are the locations to which UV radiation is administered within a test site. At least five test subsites should receive UV doses within each test site. Test subsites should be at least 0.5 square centimeters (cm
2) in area and should be separated from each other by at least 0.8 cm. Each test subsite should be outlined with indelible ink.
(iii) Applying test materials. Apply the sunscreen test product and the SPF standard at 2 milligrams per square centimeter (mg/cm
2) to their respective test sites. Use a finger cot compatible with the sunscreen to spread the product as evenly as possible.
(iv) Waiting period. Wait at least 15 minutes after applying a sunscreen product before exposing the test sites to UV radiation as described in paragraph (i)(5)) of this section. For water resistant sunscreen products, proceed with the water resistance testing procedure described in paragraph (i)(7) of this section after waiting at least 15 minutes.
(5) UV exposure - (i) Definition of minimal erythema dose (MED). The minimal erythema dose (MED) is the smallest UV dose that produces perceptible redness of the skin (erythema) with clearly defined borders at 16 to 24 hours after UV exposure. The MED for unprotected skin (MEDu) is determined on a test site that does not have sunscreen applied. The MED for protected skin (MEDp) is determined on a test site that has sunscreen applied. An MEDp is determined for the SPF standard (ssMEDp). An MEDp is determined for the sunscreen test product (tpMEDp).
(ii) UV exposure for initial MEDu. For each test subject, administer a series of UV radiation doses expressed as J/m
2-eff (as determined according to paragraph (a)(2) of this section) to the test subsites within an unprotected test site using an accurately calibrated solar simulator. Select doses that are a geometric series represented by 1.25
n (i.e., each dose is 25 percent greater than the previous dose).
(iii) UV exposure for final MEDu, ssMEDp, and tpMEDp. For each subject, determine the final MEDu, ssMEDp, and tpMEDp by administering a series of five UV doses to the appropriate test sites. The middle dose (X) in each of these dose series (i.e., the third dose) should equal the initial MEDu times the expected SPF. Note that the expected SPF equals 1 and 16.3 for the final MEDu and ssMEDp, respectively. The remaining UV doses in the series depend upon the expected SPF value of the sunscreen test product(s).
For products with an expected SPF less than 8, administer UV doses that increase by 25 percent with each successive dose (i.e., 0.64X, 0.80X, 1.00X, 1.25X, and 1.56X). For products with an expected SPF from 8 to 15, administer UV doses that increase by 20 percent with each successive dose (i.e., 0.69X, 0.83X, 1.00X, 1.20X, and 1.44X). For products with an expected SPF higher than 15, administer UV doses that increase by 15 percent with each successive dose (i.e., 0.76X, 0.87X, 1.00X, 1.15X, and 1.32X).
(iv) Evaluation of test subsites. In order that the person who evaluates the test subsites is not biased, he/she should not be the same person who applied the sunscreen drug product to the test site or administered the UV doses. After UV doses are administered, all immediate responses should be recorded. These may include an immediate darkening or tanning, typically grayish or purplish in color, which fades in 30 to 60 minutes; an immediate reddening at the subsite, due to heating of the skin, which fades rapidly; and an immediate generalized heat response, spreading beyond the subsite, which fades in 30 to 60 minutes. After the immediate responses are noted, each subject should shield the exposed area from further UV radiation until the MED is determined. Determine the MED 16 to 24 hours after UV exposure. Because erythema is evaluated 16 to 24 hours after UV exposure, the final MEDu, ssMEDp, and tpMEDp are typically determined the day following determination of the initial MEDu. Evaluate the erythema responses of each test subsite using either tungsten or warm white fluorescent lighting that provides at least 450 lux of illumination at the test site. For the evaluation, the test subject should be in the same position as when the test site was irradiated.
(v) Invalid test data. Reject test data for a test subject if erythema is not present on either the unprotected or protected test sites; or erythema is present at all subsites; or the responses are inconsistent with the series of UV doses administered; or the subject was noncompliant (e.g., the subject withdraws from the test due to illness or work conflicts or does not shield the exposed testing sites from further UV radiation until the MED is determined).
(6) Determination of SPF. (i) Calculate an SPF value for each test subject (SPFi) as follows:
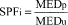
(ii) Calculate the mean
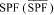
and the standard deviation (s) from the SPFi values. Calculate the standard error (SE), which equals s/â??n (where n equals the number of subjects who provided valid test results). Obtain the t value from Student's t distribution table corresponding to the upper 5-percent point with n - 1 degrees of freedom. Determine the labeled SPF value, which equals the largest whole number less than
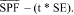
In order for the SPF determination of a test product to be considered valid, the SPF value of the SPF standard should fall within the standard deviation range of the expected SPF (i.e., 16.3 +/-3.43).
(7) Determination of water resistance. The following procedure should be performed in an indoor fresh water pool, whirlpool, and/or hot tub maintained at 23 to 32 deg.C. Fresh water is clean drinking water that meets the standards in 40 CFR part 141. The pool and air temperature and the relative humidity should be recorded.
(i) Water resistance (40 minutes). The labeled SPF should be determined after 40 minutes of water immersion using the following procedure:
(A) Step 1: Apply the sunscreen as described in paragraph (d) of this section.
(B) Step 2: Perform moderate activity in water for 20 minutes.
(C) Step 3: Rest out of water for 15 minutes. Do not towel test site(s).
(D) Step 4: Perform moderate activity in water for 20 minutes.
(E) Step 5: Allow test sites to dry completely without toweling.
(F) Step 6: Apply the SPF standard as described in paragraph (d) of this section.
Step 1. Expose test sites to UV doses as described in paragraph (e) of this section.
(ii) Water resistance (80 minutes). The labeled SPF should be determined after 80 minutes of water immersion using the following procedure:
(A) Step 1: Apply the sunscreen as described in paragraph (d) of this section.
(B) Step 2: Perform moderate activity in water for 20 minutes.
(C) Step 3: Rest out of water for 15 minutes. Do not towel test site(s).
(D) Step 4: Perform moderate activity in water for 20 minutes.
(E) Step 5: Rest out of water for 15 minutes. Do not towel test site(s).
(F) Step 6: Perform moderate activity in water for 20 minutes.
(G) Step 7: Rest out of water for 15 minutes. Do not towel test site(s).
(H) Step 8: Perform moderate activity in water for 20 minutes.
(I) Step 9: Allow test sites to dry completely without toweling.
(J) Step 10: Apply the SPF standard as described in paragraph (d) of this section.
(K) Step 11: Expose test sites to UV doses as described in paragraph (e) of this section.
(j) Broad spectrum test procedure - (1) UV Spectrometry. (i) Plate. Use optical-grade polymethylmethacrylate (PMMA) plates suitable for UV transmittance measurements. The plate should be roughened on one side to a three dimensional surface topography measure (Sa) between 2 and 7 micrometers and must have a rectangular application area of at least 16 square centimeters (with no side shorter than 4 cm).
(ii) Sample holder. The sample holder should hold the PMMA plate in a horizontal position to avoid flowing of the sunscreen drug product from one edge of the PMMA plate to the other. It should be mounted as close as possible to the input optics of the spectrometer to maximize capture of forward scattered radiation. The sample holder should be a thin, flat plate with a suitable aperture through which UV radiation can pass. The PMMA plate should be placed on the upper surface of the sample holder with the roughened side facing up.
(iii) Light source. The light source should produce a continuous spectral distribution of UV radiation from 290 to 400 nanometers.
(iv) Input optics. Unless the spectrometer is equipped with an integrating sphere, an ultraviolet radiation diffuser should be placed between the sample and the input optics of the spectrometer. The diffuser will be constructed from any UV radiation transparent material (e.g., Teflon (R) or quartz). The diffuser ensures that the radiation received by the spectrometer is not collimated. The spectrometer input slits should be set to provide a bandwidth that is less than or equal to 1 nanometer.
(v) Dynamic range of the spectrometer. The dynamic range of the spectrometer should be sufficient to measure transmittance accurately through a highly absorbing sunscreen product at all terrestrial solar UV wavelengths (290 to 400 nm).
(2) Sunscreen product application to PMMA plate. The accuracy of the test depends upon the application of a precisely controlled amount of sunscreen product with a uniform distribution over the PMMA plate. The product is applied at 0.75 mg per square centimeter to the roughened side of the PMMA plate. The sunscreen product should be applied in a series of small dots over the entire PMMA plate and then spread evenly using a gloved finger. Spreading should be done with a very light spreading action for approximately 30 seconds followed by spreading with greater pressure for approximately 30 seconds. The plate should then be allowed to equilibrate for 15 minutes in the dark before the pre-irradiation described in paragraph (c) of this section.
(3) Sunscreen product pre-irradiation. To account for lack of photostability, apply the sunscreen product to the PMMA plate as described in paragraph (b) of this section and then irradiate with a solar simulator described in section 352.70(b) of this chapter. The irradiation dose should be 4 MEDs which is equivalent to an erythemal effective dose of 800 J/m
2 (i.e., 800 J/m
2-eff).
(4) Calculation of mean transmittance values. After pre-irradiation described in paragraph (c) of this section, mean transmittance values should be determined for each wavelength λ over the full UV spectrum (290 to 400 nanometers). The transmittance values should be measured at 1 nanometer intervals. Measurements of spectral irradiance transmitted for each wavelength λ through control PMMA plates coated with 15 microliters of glycerin (no sunscreen product) should be obtained from at least 5 different locations on the PMMA plate [C1(λ), C2(λ), C3(λ), C4(λ), and C5(λ)]. In addition, a minimum of 5 measurements of spectral irradiance transmitted for each wavelength λ through the PMMA plate covered with the sunscreen product will be similarly obtained after pre-irradiation of the sunscreen product [P1(λ), P2(λ), P3(λ), P4(λ), and P5(λ)].
The mean transmittance for each wavelength,
is the ratio of the mean of the C(λ) values to the mean of the P(λ) values, as follows:
Where n â?¥5
(5) Calculation of mean absorbance values. (i) Mean transmittance values,
are converted into mean absorbance values,
at each wavelength by taking the negative logarithm of the mean transmittance value as follows:
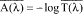
(ii) The calculation yields 111 monochromatic absorbance values in 1 nanometer increments from 290 to 400 nanometers.
(6) Number of plates. For each sunscreen product, mean absorbance values should be determined from at least three individual PMMA plates. Because paragraph (d) of this section requires at least 5 measurements per plate, there should be a total of at least 15 measurements.
(7) Calculation of the critical wavelength. The critical wavelength is identified as the wavelength at which the integral of the spectral absorbance curve reaches 90 percent of the integral over the UV spectrum from 290 to 400 nm. The following equation defines the critical wavelength:
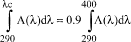
Where λc = critical wavelength
A(λ) = mean absorbance at each wavelength
dλ = wavelength interval between measurements
A mean critical wavelength of 370 nm or greater is classified as broad spectrum protection.
[76 FR 35660, June 17, 2011, as amended at 76 FR 38975, July 5, 2011]
|
|
|
Sec. 201.328 Labeling of medical gas containers.
|
|
(a) Portable cryogenic medical gas containers. For the purposes of this section a "portable cryogenic medical gas container" is one that is capable of being transported and is intended to be attached to a medical gas supply system within a hospital, health care entity, nursing home, other facility, or home health care setting, or is a base unit used to fill small cryogenic gas containers for use by individual patients. The term does not include cryogenic containers that are not designed to be connected to a medical gas supply system, e.g., tank trucks, trailers, rail cars, or small cryogenic gas containers for use by individual patients (including portable liquid oxygen units as defined at § 868.5655 of this chapter).
(1) Each portable cryogenic medical gas container must be conspicuously marked with a 360deg. wraparound label identifying its contents. Such label must meet the requirements of § 211.94(e)(2) of this chapter and the following additional requirements.
(i) If the container holds a single gas, the name of the gas held in the container must be printed on the label in one of the following ways:
(A) Using lettering that appears in the color designated for the gas in paragraph (c) of this section and that is printed against a white background, or
(B) Using lettering that appears in white against a background that is painted in the color for the gas designated in paragraph (c) of this section.
(ii) The lettering for the name of the gas on the label must be at least 2 inches high.
(iii) The name of the gas must be printed continuously around the label and be capable of being read around the entire container.
(iv) The label must be on the sidewall of the container, as close to the top of the container as possible but below the top weld seam.
(v) A portable cryogenic medical gas container may only be colored in the color or colors designated in paragraph (c) of this section if the gas or gases held within the container correspond to that color or those colors.
(2) A label on the container (either the 360deg. wraparound label required in paragraph (a)(1) of this section or a separate label) must include, in conspicuous lettering, the phrase "For Medical Use", "Medical Gas," or some similar phrase that indicates the gas is for medical use.
(b) High-pressure medical gas cylinders. Each high-pressure medical gas cylinder must be colored on the shoulder portion of the cylinder in the color or colors designated in paragraph (c) of this section. The color or colors must be visible when viewed from the top of cylinder.
(c) Medical gas colors. The colors required to identify medical gases under paragraph (a) and (b) of this section are:
| Medical gas
| Color
|
|---|
| Medical Air | Yellow.
| | Carbon Dioxide | Gray.
| | Helium | Brown.
| | Nitrogen | Black.
| | Nitrous Oxide | Blue.
| | Oxygen | Green.
| | Mixture or Blend | Colors corresponding to each component gas. |
[81 FR 81696, Nov. 18, 2016]
|
|
|
Appendix A to Part 201 - Examples of Graphic Enhancements Used by FDA
|
I. Section 201.66 Standard Labeling Format
A. Overall
1. The "Drug Facts" labeling is set off in a box or similar enclosure by the use of a barline with all black type printed on a white, color contrasting background.
B. Typeface and size
1. "Drug Facts" is set in 14 point Helvetica Bold Italic, left justified.
2. "Drug Facts (continued)" is set in 8 point Helvetica Bold Italic for the words "Drug Facts" and 8 point Helvetica Regular for the word "(continued)" and is left justified.
3. The headings (e.g., "Directions") are set in 8 point Helvetica Bold Italic, left justified.
4. The subheadings (e.g., "Ask a doctor or pharmacist before use if you are") are set in 6 point Helvetica Bold, left justified.
5. The information is set in 6 point Helvetica Regular with 6.5 point leading, left justified.
6. The heading "Purpose" is right justified.
7. The bullet is a 5-point solid square.
8. Two em spacing separates bullets when more than one bullet is on the same line.
9. A table format is used for 3 or more dosage directions.
10. A graphic appears at the bottom of the first panel leading the reader to the next panel.
C. Barlines and hairlines
1. A 2.5-point horizontal barline extends to each end of the "Drug Facts" box (or similar enclosure), providing separation between each of the headings.
2. A 0.5-point horizontal hairline extends within 2 spaces on either side of the "Drug Facts" box (or similar enclosure), immediately following the title and immediately preceding the subheadings.
3. A 0.5-point horizontal hairline follows the title, immediately preceding the heading, when a heading appears on a subsequent panel immediately after the "Drug Facts (continued)" title.
D. Box or Enclosure
1. All information is enclosed by a 2.5-point barline.
II. Section 201.66 Modified Labeling Format
A. Overall
1. The "Drug Facts" labeling is presented in all black type printed on a white color contrasting background.
B. Typeface and size
1. "Drug Facts" is set in 9 point Helvetica Bold Italic, left justified.
2. The headings (e.g., "Directions") are set in 8 point Helvetica Bold Italic, left justified.
3. The subheadings (e.g., "Ask a doctor or pharmacist before use if you are") are set in 6 point Helvetica Bold, left justified.
4. The information is set in 6 point Helvetica Regular with 6.5 point leading, left justified.
5. The heading "Purpose" is right justified.
6. The bullet is a 5-point solid square.
7. Bulleted information may start on same line as headings (except for the "Warnings" heading) and subheadings, with 2 em spacing separating bullets, and need not be vertically aligned.
C. Barlines and hairlines
1. A 2.5-point horizontal barline extends to each end of the "Drug Facts" box (or similar enclosure), providing separation between each of the headings.
2. A 0.5-point horizontal hairline extends within 2 spaces on either side of the "Drug Facts" box (or similar enclosure), immediately following the title and immediately preceding the subheadings.
D. Box or Enclosure
1. All information is set off by color contrast. No barline is used.
III. Examples of § 201.66 Standard Labeling and Modified Labeling Formats
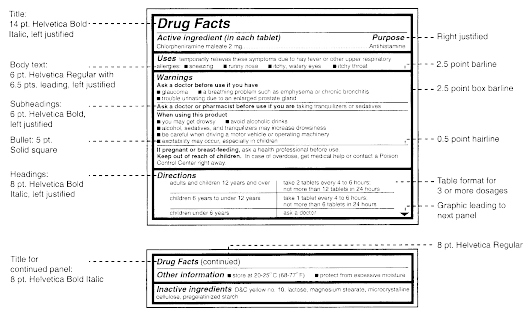
 I. Section 201.66 Standard Labeling Format
A. Overall
1. The "Drug Facts" labeling is set off in a box or similar enclosure by the use of a barline with all black type printed on a white, color contrasting background.
B. Typeface and size
1. "Drug Facts" is set in 14 point Helvetica Bold Italic, left justified.
2. "Drug Facts (continued)" is set in 8 point Helvetica Bold Italic for the words "Drug Facts" and 8 point Helvetica Regular for the word "(continued)" and is left justified.
3. The headings (e.g., "Directions") are set in 8 point Helvetica Bold Italic, left justified.
4. The subheadings (e.g., "Ask a doctor or pharmacist before use if you are") are set in 6 point Helvetica Bold, left justified.
5. The information is set in 6 point Helvetica Regular with 6.5 point leading, left justified.
6. The heading "Purpose" is right justified.
7. The bullet is a 5-point solid square.
8. Two em spacing separates bullets when more than one bullet is on the same line.
9. A table format is used for 3 or more dosage directions.
10. A graphic appears at the bottom of the first panel leading the reader to the next panel.
C. Barlines and hairlines
1. A 2.5-point horizontal barline extends to each end of the "Drug Facts" box (or similar enclosure), providing separation between each of the headings.
2. A 0.5-point horizontal hairline extends within 2 spaces on either side of the "Drug Facts" box (or similar enclosure), immediately following the title and immediately preceding the subheadings.
3. A 0.5-point horizontal hairline follows the title, immediately preceding the heading, when a heading appears on a subsequent panel immediately after the "Drug Facts (continued)" title.
D. Box or Enclosure
1. All information is enclosed by a 2.5-point barline.
II. Section 201.66 Modified Labeling Format
A. Overall
1. The "Drug Facts" labeling is presented in all black type printed on a white color contrasting background.
B. Typeface and size
1. "Drug Facts" is set in 9 point Helvetica Bold Italic, left justified.
2. The headings (e.g., "Directions") are set in 8 point Helvetica Bold Italic, left justified.
3. The subheadings (e.g., "Ask a doctor or pharmacist before use if you are") are set in 6 point Helvetica Bold, left justified.
4. The information is set in 6 point Helvetica Regular with 6.5 point leading, left justified.
5. The heading "Purpose" is right justified.
6. The bullet is a 5-point solid square.
7. Bulleted information may start on same line as headings (except for the "Warnings" heading) and subheadings, with 2 em spacing separating bullets, and need not be vertically aligned.
C. Barlines and hairlines
1. A 2.5-point horizontal barline extends to each end of the "Drug Facts" box (or similar enclosure), providing separation between each of the headings.
2. A 0.5-point horizontal hairline extends within 2 spaces on either side of the "Drug Facts" box (or similar enclosure), immediately following the title and immediately preceding the subheadings.
D. Box or Enclosure
1. All information is set off by color contrast. No barline is used.
III. Examples of § 201.66 Standard Labeling and Modified Labeling Formats
A. Section 201.66 Standard Labeling Format B. Section 201.66 Modified Labeling Format
|
|
|
Authority: 21 U.S.C. 321, 331, 343, 351, 352, 353, 355, 358, 360, 360b, 360ccc, 360ccc-1, 360ee, 360gg-360ss, 371, 374, 379e; 42 U.S.C. 216, 241, 262, 264.
Source: 40 FR 13998, Mar. 27, 1975, unless otherwise noted.
|
|





