Based on rank analysis
Return to Table of Contents
INDICATIONS AND USAGE
HUMIRA is indicated for reducing signs and symptoms and inhibiting the progression of structural damage in adult patients with moderately to severely active rheumatoid arthritis who have had an inadequate response to one or more DMARDs. HUMIRA can be used alone or in combination with MTX or other DMARDs.
CONTRAINDICATIONS
HUMIRA should not be administered to patients with known hypersensitivity to HUMIRA or any of its components.
Return to Table of Contents
WARNINGS
SERIOUS INFECTIONS AND SEPSIS, INCLUDING FATALITIES, HAVE BEEN REPORTED WITH THE USE OF TNF BLOCKING AGENTS INCLUDING HUMIRA. MANY OF THE SERIOUS INFECTIONS HAVE OCCURRED IN PATIENTS ON CONCOMITANT IMMUNOSUPPRESSIVE THERAPY THAT, IN ADDITION TO THEIR RHEUMATOID ARTHRITIS, COULD PREDISPOSE THEM TO INFECTIONS. TUBERCULOSIS AND INVASIVE OPPORTUNISTIC FUNGAL INFECTIONS HAVE BEEN OBSERVED IN PATIENTS TREATED WITH TNF BLOCKING AGENTS INCLUDING HUMIRA.
TREATMENT WITH HUMIRA SHOULD NOT BE INITIATED IN PATIENTS WITH ACTIVE INFECTIONS INCLUDING CHRONIC OR LOCALIZED INFECTIONS. PATIENTS WHO DEVELOP A NEW INFECTION WHILE UNDERGOING TREATMENT WITH HUMIRA SHOULD BE MONITORED CLOSELY. ADMINISTRATION OF HUMIRA SHOULD BE DISCONTINUED IF A PATIENT DEVELOPS A SERIOUS INFECTION. PHYSICIANS SHOULD EXERCISE CAUTION WHEN CONSIDERING THE USE OF HUMIRA IN PATIENTS WITH A HISTORY OF RECURRENT INFECTION OR UNDERLYING CONDITIONS WHICH MAY PREDISPOSE THEM TO INFECTIONS, OR PATIENTS WHO HAVE RESIDED IN REGIONS WHERE TUBERCULOSIS AND HISTOPLASMOSIS ARE ENDEMIC (see PRECAUTIONS - Tuberculosis and ADVERSE REACTIONS - Infections). THE BENEFITS AND RISKS OF HUMIRA TREATMENT SHOULD BE CAREFULLY CONSIDERED BEFORE INITIATION OF HUMIRA THERAPY.
Neurologic Events
Use of TNF blocking agents, including HUMIRA, has been associated with rare cases of exacerbation of clinical symptoms and/or radiographic evidence of demyelinating disease. Prescribers should exercise caution in considering the use of HUMIRA in patients with preexisting or recent-onset central nervous system demyelinating disorders.
Malignancies
Lymphomas have been observed in patients treated with TNF blocking agents including HUMIRA. In clinical trials, patients treated with HUMIRA had a higher incidence of lymphoma than the expected rate in the general population (see ADVERSE REACTIONS-Malignancies). While patients with rheumatoid arthritis, particularly those with highly active disease, may be at a higher risk (up to several fold) for the development of lymphoma, the role of TNF blockers in the development of malignancy is not known4,5.
Return to Table of Contents
PRECAUTIONS
General
Allergic reactions have been observed in approximately 1% of patients receiving HUMIRA. If an anaphylactic reaction or other serious allergic reaction occurs, administration of HUMIRA should be discontinued immediately and appropriate therapy initiated.
Information to Patients
The first injection should be performed under the supervision of a qualified health care professional. If a patient or caregiver is to administer HUMIRA, he/she should be instructed in injection techniques and their ability to inject subcutaneously should be assessed to ensure the proper administration of HUMIRA (see HUMIRA, PATIENT INFORMATION LEAFLET). A puncture-resistant container for disposal of needles and syringes should be used. Patients or caregivers should be instructed in the technique as well as proper syringe and needle disposal, and be cautioned against reuse of these items.
Tuberculosis
As observed with other TNF blocking agents, tuberculosis associated with the administration of HUMIRA in clinical trials has been reported (see WARNINGS). While cases were observed at all doses, the incidence of tuberculosis reactivations was particularly increased at doses of HUMIRA that were higher than the recommended dose. All patients recovered after standard antimicrobial therapy. No deaths due to tuberculosis occurred during the clinical trials.
Before initiation of therapy with HUMIRA, patients should be evaluated for active or latent tuberculosis infection with a tuberculin skin test. If latent infection is diagnosed, appropriate prophylaxis in accordance with the Centers for Disease Control and Prevention guidelines6 should be instituted. Patients should be instructed to seek medical advice if signs/symptoms (e.g., persistent cough, wasting/weight loss, low grade fever) suggestive of a tuberculosis infection occur.
Immunosuppression
The possibility exists for TNF blocking agents, including HUMIRA, to affect host defenses against infections and malignancies since TNF mediates inflammation and modulates cellular immune responses. In a study of 64 patients with rheumatoid arthritis treated with HUMIRA, there was no evidence of depression of delayed-type hypersensitivity, depression of immunoglobulin levels, or change in enumeration of effector T- and B-cells and NK-cells, monocyte/macrophages, and neutrophils. The impact of treatment with HUMIRA on the development and course of malignancies, as well as active and/or chronic infections is not fully understood (see WARNINGS, ADVERSE REACTIONS, Infections and Malignancies). The safety and efficacy of HUMIRA in patients with immunosuppression have not been evaluated.
Immunizations
No data are available on the effects of vaccination in patients receiving HUMIRA. Live vaccines should not be given concurrently with HUMIRA. No data are available on the secondary transmission of infection by live vaccines in patients receiving HUMIRA.
Autoimmunity
Treatment with HUMIRA may result in the formation of autoantibodies and, rarely, in the development of a lupus-like syndrome. If a patient develops symptoms suggestive of a lupus-like syndrome following treatment with HUMIRA, treatment should be discontinued (see ADVERSE REACTIONS, Autoantibodies).
Drug Interactions
HUMIRA has been studied in rheumatoid arthritis patients taking concomitant MTX (see CLINICAL PHARMACOLOGY: Drug Interactions). The data do not suggest the need for dose adjustment of either HUMIRA or MTX.
Carcinogenesis, Mutagenesis, and Impairment of Fertility
Long-term animal studies of HUMIRA have not been conducted to evaluate the carcinogenic potential or its effect on fertility. No clastogenic or mutagenic effects of HUMIRA were observed in the in vivo mouse micronucleus test or the Salmonella-Escherichia coli (Ames) assay, respectively.
Pregnancy
Pregnancy Category B - An embryo-fetal perinatal developmental toxicity study has been performed in cynomolgus monkeys at dosages up to 100 mg/kg (266 times human AUC when given 40 mg subcutaneous with MTX every week or 373 times human AUC when given 40 mg subcutaneous without MTX) and has revealed no evidence of harm to the fetuses due to adalimumab. There are, however, no adequate and well-controlled studies in pregnant women. Because animal reproduction and developmental studies are not always predictive of human response, HUMIRA should be used during pregnancy only if clearly needed.
Nursing Mothers
It is not known whether adalimumab is excreted in human milk or absorbed systemically after ingestion. Because many drugs and immunoglobulins are excreted in human milk, and because of the potential for serious adverse reactions in nursing infants from HUMIRA, a decision should be made whether to discontinue nursing or to discontinue the drug, taking into account the importance of the drug to the mother.
Pediatric Use
Safety and effectiveness of HUMIRA in pediatric patients have not been established.
Geriatric Use
A total of 519 patients 65 years of age and older, including 107 patients 75 years and older, received HUMIRA in clinical studies. No overall difference in effectiveness was observed between these subjects and younger subjects. The frequency of serious infection and malignancy among HUMIRA treated subjects over age 65 was higher than for those under age 65. Because there is a higher incidence of infections and malignancies in the elderly population in general, caution should be used when treating the elderly.
Return to Table of Contents
ADVERSE REACTIONS
General
The most serious adverse reactions were (see WARNINGS):
- Serious Infections
- Neurologic Events
- Malignancies
The most common adverse reaction with HUMIRA was injection site reactions. In placebo-controlled trials, 20% of patients treated with HUMIRA developed injection site reactions (erythema and/or itching, hemorrhage, pain or swelling), compared to 14% of patients receiving placebo. Most injection site reactions were described as mild and generally did not necessitate drug discontinuation.
The proportion of patients who discontinued treatment due to adverse events during the double-blind, placebo-controlled portion of Studies I, II, III and IV was 7% for patients taking HUMIRA and 4% for placebo-treated patients. The most common adverse events leading to discontinuation of HUMIRA were clinical flare reaction (0.7%), rash (0.3%) and pneumonia (0.3%).
Because clinical trials are conducted under widely varying and controlled conditions, adverse reaction rates observed in clinical trials of a drug cannot be directly compared to rates in the clinical trials of another drug and may not predict the rates observed in a broader patient population in clinical practice.
Infections
In placebo-controlled trials, the rate of infection was 1 per patient year in the HUMIRA treated patients and 0.9 per patient year in the placebo-treated patients. The infections consisted primarily of upper respiratory tract infections, bronchitis and urinary tract infections. Most patients continued on HUMIRA after the infection resolved. The incidence of serious infections was 0.04 per patient year in HUMIRA treated patients and 0.02 per patient year in placebo-treated patients. Serious infections observed included pneumonia, septic arthritis, prosthetic and post-surgical infections, erysipelas, cellulitis, diverticulitis, and pyelonephritis (see WARNINGS).
Thirteen cases of tuberculosis, including miliary, lymphatic, peritoneal, and pulmonary were reported in clinical trials. Most of the cases of tuberculosis occurred within the first eight months after initiation of therapy and may reflect recrudescence of latent disease. Six cases of invasive opportunistic infections caused by histoplasma, aspergillus, and nocardia were also reported in clinical trials (see WARNINGS).
Malignancies
Among 2468 rheumatoid arthritis patients treated in clinical trials with HUMIRA for a median of 24 months, 48 malignancies of various types were observed, including 10 patients with lymphoma. The Standardized Incidence Ratio (SIR) (ratio of observed rate to age-adjusted expected frequency in the general population) for malignancies was 1.0 (95% CI, 0.7, 1.3) and for lymphomas was 5.4 (95% CI, 2.6, 10.0). An increase of up to several fold in the rate of lymphomas has been reported in the rheumatoid arthritis patient population4, and may be further increased in patients with more severe disease activity5 (see WARNINGS-Malignancies). The other malignancies observed during use of HUMIRA were breast, colon-rectum, uterine-cervical, prostate, melanoma, gallbladder-bile ducts, and other carcinomas.
Autoantibodies
In the controlled trials, 12% of patients treated with HUMIRA and 7% of placebo-treated patients that had negative baseline ANA titers developed positive titers at week 24. One patient out of 2334 treated with HUMIRA developed clinical signs suggestive of new-onset lupus-like syndrome. The patient improved following discontinuation of therapy. No patients developed lupus nephritis or central nervous system symptoms. The impact of long-term treatment with HUMIRA on the development of autoimmune diseases is unknown.
Immunogenicity
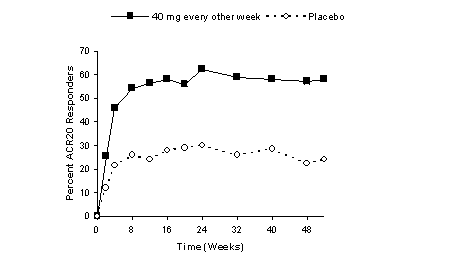
Patients in Studies I, II, and III were tested at multiple time points for antibodies to adalimumab during the 6 to 12 month period. Approximately 5% (58 of 1,062) of adult rheumatoid arthritis patients receiving HUMIRA developed low-titer antibodies to adalimumab at least once during treatment, which were neutralizing in vitro. Patients treated with concomitant MTX had a lower rate of antibody development than patients on HUMIRA monotherapy (1% versus 12%). No apparent correlation of antibody development to adverse events was observed. With monotherapy, patients receiving every other week dosing may develop antibodies more frequently than those receiving weekly dosing. In patients receiving the recommended dosage of 40 mg every other week as monotherapy, the ACR 20 response was lower among antibody-positive patients than among antibody-negative patients. The long-term immunogenicity of HUMIRA is unknown.
The data reflect the percentage of patients whose test results were considered positive for antibodies to adalimumab in an ELISA assay, and are highly dependent on the sensitivity and specificity of the assay. Additionally the observed incidence of antibody positivity in an assay may be influenced by several factors including sample handling, timing of sample collection, concomitant medications, and underlying disease. For these reasons, comparison of the incidence of antibodies to adalimumab with the incidence of antibodies to other products may be misleading.
Other Adverse Reactions
The data described below reflect exposure to HUMIRA in 2334 patients, including 2073 exposed for 6 months, 1497 exposed for greater than one year and 1380 in adequate and well-controlled studies (Studies I, II, III, and IV). HUMIRA was studied primarily in placebo-controlled trials and in long-term follow up studies for up to 36 months duration. The population had a mean age of 54 years, 77% were female, 91% were Caucasian and had moderately to severely active rheumatoid arthritis. Most patients received 40 mg HUMIRA every other week.
Table 4 summarizes events reported at a rate of at least 5% in patients treated with HUMIRA 40 mg every other week compared to placebo and with an incidence higher than placebo. Adverse event rates in patients treated with HUMIRA 40 mg weekly were similar to rates in patients treated with HUMIRA 40 mg every other week.
Table 4: Adverse Events Reported by > 5% of Patients Treated with HUMIRA During Placebo-Controlled Period of Rheumatoid Arthritis Studies
| |
HUMIRA
40 mg subcutaneous
Every Other Week
(N=705) |
Placebo
(N=690) |
| Adverse Event (Preferred Term) |
Percentage |
Percentage |
| Respiratory |
| Upper respiratory infection |
17 |
13 |
| Sinusitis |
11 |
9 |
| Flu syndrome |
7 |
6 |
| Gastrointestinal |
| Nausea |
9 |
8 |
| Abdominal pain |
7 |
4 |
| Laboratory Tests* |
| Laboratory test abnormal |
8 |
7 |
| Hypercholesterolemia |
6 |
4 |
| Hyperlipidemia |
7 |
5 |
| Hematuria |
5 |
4 |
| Alkaline phosphatase increased |
5 |
3 |
| Other |
| Injection site pain |
12 |
12 |
| Headache |
12 |
8 |
| Rash |
12 |
6 |
| Accidental injury |
10 |
8 |
| Injection site reaction** |
8 |
1 |
| Back pain |
6 |
4 |
| Urinary tract infection |
8 |
5 |
| Hypertension |
5 |
3 |
* Laboratory test abnormalities were reported as adverse events in European trials
** Does not include erythema and/or itching, hemorrhage, pain or swelling
Other Adverse Events
Other infrequent serious adverse events occurring at an incidence of less than 5% in patients treated with HUMIRA were:
Body As A Whole: Fever, infection, pain in extremity, pelvic pain, sepsis, surgery, thorax pain, tuberculosis reactivated
Cardiovascular System: Arrhythmia, atrial fibrillation, cardiovascular disorder, chest pain, congestive heart failure, coronary artery disorder, heart arrest, hypertensive encephalopathy, myocardial infarct, palpitation, pericardial effusion, pericarditis, syncope, tachycardia, vascular disorder
Collagen Disorder: Lupus erythematosus syndrome
Digestive System: Cholecystitis, cholelithiasis, esophagitis, gastroenteritis, gastrointestinal disorder, gastrointestinal hemorrhage, hepatic necrosis, vomiting
Endocrine System: Parathyroid disorder
Hemic And Lymphatic System: Agranulocytosis, granulocytopenia, leukopenia, lymphoma like reaction, pancytopenia, polycythemia
Metabolic And Nutritional Disorders: Dehydration, healing abnormal, ketosis, paraproteinemia, peripheral edema
Musculo-Skeletal System: Arthritis, bone disorder, bone fracture (not spontaneous), bone necrosis, joint disorder, muscle cramps, myasthenia, pyogenic arthritis, synovitis, tendon disorder
Neoplasia: Adenoma, carcinomas such as breast, gastrointestinal, skin, urogenital, and others; lymphoma and melanoma.
Nervous System: Confusion, multiple sclerosis, paresthesia, subdural hematoma, tremor
Respiratory System: Asthma, bronchospasm, dyspnea, lung disorder, lung function decreased, pleural effusion, pneumonia
Skin And Appendages: Cellulitis, erysipelas, herpes zoster
Special Senses: Cataract
Thrombosis: Thrombosis leg
Urogenital System: Cystitis, kidney calculus, menstrual disorder, pyelonephritis
Return to Table of Contents
OVERDOSAGE
The maximum tolerated dose of HUMIRA has not been established in humans. Multiple doses up to 10 mg/kg have been administered to patients in clinical trials without evidence of dose-limiting toxicities. In case of overdosage, it is recommended that the patient be monitored for any signs or symptoms of adverse reactions or effects and appropriate symptomatic treatment instituted immediately.
Return to Table of Contents
DOSAGE AND ADMINISTRATION
The recommended dose of HUMIRA for adult patients with rheumatoid arthritis is 40 mg administered every other week as a subcutaneous injection. MTX, glucocorticoids, salicylates, nonsteroidal anti-inflammatory drugs (NSAIDs), analgesics or other DMARDs may be continued during treatment with HUMIRA. Some patients not taking concomitant MTX may derive additional benefit from increasing the dosing frequency of HUMIRA to 40 mg every week.
HUMIRA is intended for use under the guidance and supervision of a physician. Patients may self-inject HUMIRA if their physician determines that it is appropriate and with medical follow-up, as necessary, after proper training in injection technique.
The solution in the syringe and in the vial should be carefully inspected visually for particulate matter and discoloration prior to subcutaneous administration. If particulates and discolorations are noted, the product should not be used. HUMIRA does not contain preservatives; therefore, unused portions of drug remaining from the syringe or vial should be discarded. NOTE: The needle cover of the syringe contains dry rubber (latex), which should not be handled by persons sensitive to this substance.
Patients using the pre-filled syringes should be instructed to inject the full amount in the syringe (0.8 mL), which provides 40 mg of HUMIRA. For patients and institutions using vials, 0.8 mL of solution providing 40 mg of HUMIRA should be withdrawn from the vial and administered according to the directions provided in the Patient Information Leaflet.
Injection sites should be rotated and injections should never be given into areas where the skin is tender, bruised, red or hard (see PATIENT INFORMATION LEAFLET).
Instructions For Activating the Needle Stick Device: Cartons for institutional use contain a syringe and needle with a needle protection device (see HOW SUPPLIED). To activate the needle stick protection device after injection, hold the syringe in one hand and, with the other hand, slide the outer protective shield over the exposed needle until it locks into place.
Storage and Stability
Do not use beyond the expiration date on the container. HUMIRA must be refrigerated at 2-8° C (36-46° F). DO NOT FREEZE. Protect the vial or/and pre-filled syringe from exposure to light. Store in original carton until time of administration.
Return to Table of Contents
HOW SUPPLIED
HUMIRAä (adalimumab) is supplied in glass vials and syringes as a preservative-free, sterile solution for subcutaneous administration. The following packaging configurations are available:
Patient Use Syringe Carton
HUMIRA is dispensed in a carton containing two alcohol preps and two dose trays. Each dose tray consists of a single-use, 1 mL pre-filled glass syringe with a fixed 27 gauge ˝ inch needle, providing 40 mg (0.8 mL) of HUMIRA. The NDC number is 0074-3799-02.
Patient Use Vial Carton
HUMIRA is dispensed in a carton containing four alcohol preps and two trays. Each dose tray consists of a single-use, 2 mL glass vial providing 40 mg (0.8 mL) of HUMIRA and one sterile 1 mL syringe with a fixed 25 gauge 5/8 inch needle. The NDC number is 0074-3797-02.
Institutional Use Syringe Carton
Each carton contains two alcohol preps and one tray. Each dose tray consists of a single use, 1 mL pre-filled glass syringe with a fixed 27 gauge ˝ inch needle (with a needle stick protection device) providing 40 mg (0.8 mL) HUMIRA. The NDC number is 0074-3799-01.
Institutional Use Vial Carton
Each carton contains two alcohol preps and one tray. Each dose tray consists of a 2 mL glass vial providing 40 mg (0.8 mL) of HUMIRA, and one sterile syringe with a fixed 27 gauge ˝ inch needle (with needle stick protection device). The NDC number is 0074-3797-01.
Return to Table of Contents
REFERENCES
- Arnett FC, Edworthy SM, Bloch DA, et. al. The American Rheumatology Association 1987 Revised Criteria for the Classification of Rheumatoid Arthritis. Arthritis Rheum 1988; 31:315-24.
- Ramey DR, Fries JF, Singh G. The Health Assessment Questionnaire 1995 - Status and Review. In: Spilker B, ed. “Quality of Life and Pharmacoeconomics in Clinical Trials.” 2nd ed. Philadelphia, PA. Lippincott-Raven 1996.
- Ware JE, Gandek B. Overview of the SF-36 Health Survey and the International Quality of Life Assessment (IQOLA) Project. J Clin Epidemiol 1998; 51(11):903-12.
- Mellemkjaer L, Linet MS, Gridley G, et al. Rheumatiod Arthritis and Cancer Risk. European Journal of Cancer 1996; 32A (10): 1753-1757.
- Baecklund E, Ekbom A, Sparen P, et al. Disease Activity and Risk of Lymphoma in Patients With Rheumatoid Arthritis: Nested Case-Control Study. BMJ 1998; 317: 180-181.
- Centers for Disease Control and Prevention. Targeted Tuberculin Testing and Treatment of Latent Tuberculosis Infection. MMWR 2000; 49(No. RR-6):26-38.
Issued: December 2002
Abbott Laboratories
North Chicago, IL 60064, U.S.A.
U.S. Govt. Lic. No. 0043
Return to Table of Contents
Last Updated: 1/27/2003
Date created: September 26, 2003