|
|
| [Code of Federal Regulations] |
| [Title 21, Volume 8] |
| [CITE: 21CFR862] |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES
SUBCHAPTER H - MEDICAL DEVICES
| |
| PART 862 | CLINICAL CHEMISTRY AND CLINICAL TOXICOLOGY DEVICES |
|
|
|
|
Subpart B - Clinical Chemistry Test Systems
|
|
Sec. 862.1020 Acid phosphatase (total or prostatic) test system.
|
|
(a) Identification. An acid phosphatase (total or prostatic) test system is a device intended to measure the activity of the acid phosphatase enzyme in plasma and serum.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1025 Adrenocorticotropic hormone (ACTH) test system.
|
|
(a) Identification. An adrenocorticotropic hormone (ACTH) test system is a device intended to measure adrenocorticotropic hormone in plasma and serum. ACTH measurements are used in the differential diagnosis and treatment of certain disorders of the adrenal glands such as Cushing's syndrome, adrenocortical insufficiency, and the ectopic ACTH syndrome.
(b) Classification. Class II.
|
|
|
Sec. 862.1030 Alanine amino transferase (ALT/SGPT) test system.
|
|
(a) Identification. An alanine amino transferase (ALT/SGPT) test system is a device intended to measure the activity of the enzyme alanine amino transferase (ALT) (also known as a serum glutamic pyruvic transaminase or SGPT) in serum and plasma. Alanine amino transferase measurements are used in the diagnosis and treatment of certain liver diseases (e.g., viral hepatitis and cirrhosis) and heart diseases.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1035 Albumin test system.
|
|
(a) Identification. An albumin test system is a device intended to measure the albumin concentration in serum and plasma. Albumin measurements are used in the diagnosis and treatment of numerous diseases involving primarily the liver or kidneys.
(b) Classification. Class II.
|
|
|
Sec. 862.1040 Aldolase test system.
|
|
(a) Identification. An aldolase test system is a device intended to measure the activity of the enzyme aldolase in serum or plasma. Aldolase measurements are used in the diagnosis and treatment of the early stages of acute hepatitis and for certain muscle diseases such as progressive Duchenne-type muscular dystrophy.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1045 Aldosterone test system.
|
|
(a) Identification. An aldosterone test system is a device intended to measure the hormone aldosterone in serum and urine. Aldosterone measurements are used in the diagnosis and treatment of primary aldosteronism (a disorder caused by the excessive secretion of aldosterone by the adrenal gland), hypertension caused by primary aldosteronism, selective hypoaldosteronism, edematous states, and other conditions of electrolyte imbalance.
(b) Classification. Class II.
|
|
|
Sec. 862.1050 Alkaline phosphatase or isoenzymes test system.
|
|
(a) Identification. An alkaline phosphatase or isoenzymes test system is a device intended to measure alkaline phosphatase or its isoenzymes (a group of enzymes with similar biological activity) in serum or plasma. Measurements of alkaline phosphatase or its isoenzymes are used in the diagnosis and treatment of liver, bone, parathyroid, and intestinal diseases.
(b) Classification. Class II.
|
|
|
Sec. 862.1055 Newborn screening test system for amino acids, free carnitine, and acylcarnitines using tandem mass spectrometry.
|
|
(a) Identification. A newborn screening test system for amino acids, free carnitine, and acylcarnitines using tandem mass spectrometry is a device that consists of stable isotope internal standards, control materials, extraction solutions, flow solvents, instrumentation, software packages, and other reagents and materials. The device is intended for the measurement and evaluation of amino acids, free carnitine, and acylcarnitine concentrations from newborn whole blood filter paper samples. The quantitative analysis of amino acids, free carnitine, and acylcarnitines and their relationship with each other provides analyte concentration profiles that may aid in screening newborns for one or more inborn errors of amino acid, free carnitine, and acyl-carnitine metabolism.
(b) Classification. Class II (special controls). The special control is FDA's guidance document entitled "Class II Special Controls Guidance Document: Newborn Screening Test Systems for Amino Acids, Free Carnitine, and Acylcarnitines Using Tandem Mass Spectrometry." See § 862.1(d) for the availability of this guidance document.
[69 FR 68255, Nov. 24, 2004]
|
|
|
Sec. 862.1060 Delta-aminolevulinic acid test system.
|
|
(a) Identification. A delta -aminolevulinic acid test system is a device intended to measure the level of delta -aminolevulinic acid (a precursor of porphyrin) in urine. Delta -aminolevulinic acid measurements are used in the diagnosis and treatment of lead poisoning and certain porphyrias (diseases affecting the liver, gastrointestinal, and nervous systems that are accompanied by increased urinary excretion of various heme compounds including delta -aminolevulinic acid).
(b) Classification. Class I (general controls). The device is exempt from premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1065 Ammonia test system.
|
|
(a) Identification. An ammonia test system is a device intended to measure ammonia levels in blood, serum, and plasma, Ammonia measurements are used in the diagnosis and treatment of severe liver disorders, such as cirrhosis, hepatitis, and Reye's syndrome.
(b) Classification. Class I.
|
|
|
Sec. 862.1070 Amylase test system.
|
|
(a) Identification. An amylase test system is a device intended to measure the activity of the enzyme amylase in serum and urine. Amylase measurements are used primarily for the diagnosis and treatment of pancreatitis (inflammation of the pancreas).
(b) Classification. Class II.
|
|
|
Sec. 862.1075 Androstenedione test system.
|
|
(a) Identification. An androstenedione test system is a device intended to measure androstenedione (a substance secreted by the testes, ovary, and adrenal glands) in serum. Adrostenedione measurements are used in the diagnosis and treatment of females with excessive levels of androgen (male sex hormone) production.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1080 Androsterone test system.
|
|
(a) Identification. An androsterone test system is a device intended to measure the hormone adrosterone in serum, plasma, and urine. Androsterone measurements are used in the diagnosis and treatment of gonadal and adrenal diseases.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1085 Angiotensin I and renin test system.
|
|
(a) Identification. An angiotensin I and renin test system is a device intended to measure the level of angiotensin I generated by renin in plasma. Angiotensin I measurements are used in the diagnosis and treatment of certain types of hypertension.
(b) Classification. Class II.
|
|
|
Sec. 862.1090 Angiotensin converting enzyme (A.C.E.) test system.
|
|
(a) Identification. An angiotensin converting enzyme (A.C.E.) test system is a device intended to measure the activity of angiotensin converting enzyme in serum and plasma. Measurements obtained by this device are used in the diagnosis and treatment of diseases such as sarcoidosis, a disease characterized by the formation of nodules in the lungs, bones, and skin, and Gaucher's disease, a hereditary disorder affecting the spleen.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1095 Ascorbic acid test system.
|
|
(a) Identification. An ascorbic acid test system is a device intended to measure the level of ascorbic acid (vitamin C) in plasma, serum, and urine. Ascorbic acid measurements are used in the diagnosis and treatment of ascorbic acid dietary deficiencies.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1100 Aspartate amino transferase (AST/SGOT) test system.
|
|
(a) Identification. An aspartate amino transferase (AST/SGOT) test system is a device intended to measure the activity of the enzyme aspartate amino transferase (AST) (also known as a serum glutamic oxaloacetic transferase or SGOT) in serum and plasma. Aspartate amino transferase measurements are used in the diagnosis and treatment of certain types of liver and heart disease.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1110 Bilirubin (total or direct) test system.
|
|
(a) Identification. A bilirubin (total or direct) test system is a device intended to measure the levels of bilirubin (total or direct) in plasma or serum. Measurements of the levels of bilirubin, an organic compound formed during the normal and abnormal distruction of red blood cells, if used in the diagnosis and treatment of liver, hemolytic hematological, and metabolic disorders, including hepatitis and gall bladder block.
(b) Classification. Class II.
|
|
|
Sec. 862.1113 Bilirubin (total and unbound) in the neonate test system.
|
|
(a) Identification. A bilirubin (total and unbound) in the neonate test system is a device intended to measure the levels of bilirubin (total and unbound) in the blood (serum) of newborn infants to aid in indicating the risk of bilirubin encephalopathy (kernicterus).
(b) Classification. Class I.
[54 FR 30206, July 19, 1989]
|
|
|
Sec. 862.1115 Urinary bilirubin and its conjugates (nonquantitative) test system.
|
|
(a) Identification. A urinary bilirubin and its conjugates (nonquantitative) test system is a device intended to measure the levels of bilirubin conjugates in urine. Measurements of urinary bilirubin and its conjugates (nonquantitative) are used in the diagnosis and treatment of certain liver diseases.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1117 B-type natriuretic peptide test system.
|
|
(a) Identification. The B-type natriuretic peptide (BNP) test system is an in vitro diagnostic device intended to measure BNP in whole blood and plasma. Measurements of BNP are used as an aid in the diagnosis of patients with congestive heart failure.
(b) Classification. Class II (special controls). The special control is "Class II Special Control Guidance Document for B-Type Natriuretic Peptide Premarket Notifications; Final Guidance for Industry and FDA Reviewers."
[66 FR 12734, Feb. 28, 2001]
|
|
|
Sec. 862.1118 Biotinidase test system.
|
|
(a) Identification. The biotinidase test system is an in vitro diagnostic device intended to measure the activity of the enzyme biotinidase in blood. Measurements of biotinidase are used in the treatment and diagnosis of biotinidase deficiency, an inborn error of metabolism in infants, characterized by the inability to utilize dietary protein bound vitamin or to recycle endogenous biotin. The deficiency may result in irreversible neurological impairment.
(b) Classification. Class II (special controls). The special control is sale, distribution, and use in accordance with the prescription device requirements in § 801.109 of this chapter.
[65 FR 16521, Mar. 29, 2000]
|
|
|
Sec. 862.1120 Blood gases (PCO2, PO2) and blood pH test system.
|
|
(a) Identification. A blood gases (PCO2, PO2) and blood pH test system is a device intended to measure certain gases in blood, serum, plasma or pH of blood, serum, and plasma. Measurements of blood gases (PCO2, PO2) and blood pH are used in the diagnosis and treatment of life-threatening acid-base disturbances.
(b) Classification. Class II.
|
|
|
Sec. 862.1130 Blood volume test system.
|
|
(a) Identification. A blood volume test system is a device intended to measure the circulating blood volume. Blood volume measurements are used in the diagnosis and treatment of shock, hemorrhage, and polycythemia vera (a disease characterized by an absolute increase in erythrocyte mass and total blood volume).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1135 C-peptides of proinsulin test system.
|
|
(a) Identification. A C-peptides of proinsulin test system is a device intended to measure C-peptides of proinsulin levels in serum, plasma, and urine. Measurements of C-peptides of proinsulin are used in the diagnosis and treatment of patients with abnormal insulin secretion, including diabetes mellitus.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1140 Calcitonin test system.
|
|
(a) Identification. A calcitonin test system is a device intended to measure the thyroid hormone calcitonin (thyrocalcitonin) levels in plasma and serum. Calcitonin measurements are used in the diagnosis and treatment of diseases involving the thyroid and parathyroid glands, including carcinoma and hyperparathyroidism (excessive activity of the parathyroid gland).
(b) Classification. Class II.
|
|
|
Sec. 862.1145 Calcium test system.
|
|
(a) Identification. A calcium test system is a device intended to measure the total calcium level in serum. Calcium measurements are used in the diagnosis and treatment of parathyroid disease, a variety of bone diseases, chronic renal disease and tetany (intermittent muscular contractions or spasms).
(b) Classification. Class II.
|
|
|
Sec. 862.1150 Calibrator.
|
|
(a) Identification. A calibrator is a device intended for medical purposes for use in a test system to establish points of reference that are used in the determination of values in the measurement of substances in human specimens. (See also § 862.2 in this part.)
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1155 Human chorionic gonadotropin (HCG) test system.
|
|
(a) Human chorionic gonadotropin (HCG) test system intended for the early detection of pregnancy - (1) Identification. A human chorionic gonadotropin (HCG) test system is a device intended for the early detection of pregnancy is intended to measure HCG, a placental hormone, in plasma or urine.
(2) Classification. Class II.
(b) Human chorionic gonadotropin (HCG) test system intended for any uses other than early detection of pregnancy - (1) Identification. A human chorionic goadotropin (HCG) test system is a device intended for any uses other than early detection of pregnancy (such as an aid in the diagnosis, prognosis, and management of treatment of persons with certain tumors or carcinomas) is intended to measure HCG, a placental hormone, in plasma or urine.
(2) Classification. Class III.
(3) Date PMA or notice of completion of a PDP is required. As of the enactment date of the amendments, May 28, 1976, an approval under section 515 of the act is required before the device described in paragraph (b)(1) may be commercially distributed. See § 862.3.
|
|
|
Sec. 862.1160 Bicarbonate/carbon dioxide test system.
|
|
(a) Identification. A bicarbonate/carbon dioxide test system is a device intended to measure bicarbonate/carbon dioxide in plasma, serum, and whole blood. Bicarbonate/carbon dioxide measurements are used in the diagnosis and treatment of numerous potentially serious disorders associated with changes in body acid-base balance.
(b) Classification. Class II.
|
|
|
Sec. 862.1163 Cardiac allograft gene expression profiling test system.
|
|
(a) Identification. A cardiac allograft gene expression profiling test system is a device that measures the ribonucleic acid (RNA) expression level of multiple genes and combines this information to yield a signature (pattern, classifier, index, score) to aid in the identification of a low probability of acute cellular rejection (ACR) in heart transplant recipients with stable allograft function.
(b) Classification. Class II (special controls). The special control is FDA's guidance document entitled "Class II Special Controls Guidance Document: Cardiac Allograft Gene Expression Profiling Test Systems." See § 862.1(d) for the availability of this guidance document.
[74 FR 53885, Oct. 21, 2009]
|
|
|
Sec. 862.1165 Catecholamines (total) test system.
|
|
(a) Identification. A catecholamines (total) test system is a device intended to determine whether a group of similar compounds (epinephrine, norepinephrine, and dopamine) are present in urine and plasma. Catecholamine determinations are used in the diagnosis and treatment of adrenal medulla and hypertensive disorders, and for catecholamine-secreting tumors (pheochromo-cytoma, neuroblastoma, ganglioneuroma, and retinoblastoma).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1170 Chloride test system.
|
|
(a) Identification. A chloride test system is a device intended to measure the level of chloride in plasma, serum, sweat, and urine. Chloride measurements are used in the diagnosis and treatment of electrolyte and metabolic disorders such as cystic fibrosis and diabetic acidosis.
(b) Classification. Class II.
|
|
|
Sec. 862.1175 Cholesterol (total) test system.
|
|
(a) Identification. A cholesterol (total) test system is a device intended to measure cholesterol in plasma and serum. Cholesterol measurements are used in the diagnosis and treatment of disorders involving excess cholesterol in the blood and lipid and lipoprotein metabolism disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1177 Cholylglycine test system.
|
|
(a) Identification. A cholylglycine test system is a device intended to measure the bile acid cholylglycine in serum. Measurements obtained by this device are used in the diagnosis and treatment of liver disorders, such as cirrhosis or obstructive liver disease.
(b) Classification. Class II.
|
|
|
Sec. 862.1180 Chymotrypsin test system.
|
|
(a) Identification. A chymotrypsin test system is a device intended to measure the activity of the enzyme chymotrypsin in blood and other body fluids and in feces. Chymotrypsin measurements are used in the diagnosis and treatment of pancreatic exocrine insufficiency.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1185 Compound S (11-deoxycortisol) test system.
|
|
(a) Identification. A compound S (11-dioxycortisol) test system is a device intended to measure the level of compound S (11-dioxycortisol) in plasma. Compound S is a steroid intermediate in the biosynthesis of the adrenal hormone cortisol. Measurements of compound S are used in the diagnosis and treatment of certain adrenal and pituitary gland disorders resulting in clinical symptoms of masculinization and hypertension.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1187 Conjugated sulfolithocholic acid (SLCG) test system.
|
|
(a) Identification. A conjugated sulfolithocholic acid (SLCG) test system is a device intended to measure the bile acid SLCG in serum. Measurements obtained by this device are used in the diagnosis and treatment of liver disorders, such as cirrhosis or obstructive liver disease.
(b) Classification. Class II.
|
|
|
Sec. 862.1190 Copper test system.
|
|
(a) Identification. A copper test system is a device intended to measure copper levels in plasma, serum, and urine. Measurements of copper are used in the diagnosis and treatment of anemia, infections, inflammations, and Wilson's disease (a hereditary disease primarily of the liver and nervous system). Test results are also used in monitoring patients with Hodgkin's disease (a disease primarily of the lymph system).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38787, July 25, 2001]
|
|
|
Sec. 862.1195 Corticoids test system.
|
|
(a) Identification. A corticoids test system is a device intended to measure the levels of corticoids (hormones of the adrenal cortex) in serum and p lasma. Measurements of corticoids are used in the diagnosis and treatment of disorders of the cortex of the adrenal glands, especially those associated with hypertension and electrolyte disturbances.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1200 Corticosterone test system.
|
|
(a) Identification. A corticosterone test system is a device intended to measure corticosterone (a steroid secreted by the adrenal gland) levels in plasma. Measurements of corticosterone are used in the diagnosis and treatment of adrenal disorders such as adrenal cortex disorders and blocks in cortisol synthesis.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1205 Cortisol (hydrocortisone and hydroxycorticosterone) test system.
|
|
(a) Identification. A cortisol (hydrocortisone and hydroxycorticosterone) test system is a device intended to measure the cortisol hormones secreted by the adrenal gland in plasma and urine. Measurements of cortisol are used in the diagnosis and treatment of disorders of the adrenal gland.
(b) Classification. Class II.
|
|
|
Sec. 862.1210 Creatine test system.
|
|
(a) Identification. A creatine test system is a device intended to measure creatine (a substance synthesized in the liver and pancreas and found in biological fluids) in plasma, serum, and urine. Measurements of creatine are used in the diagnosis and treatment of muscle diseases and endocrine disorders including hyperthyroidism.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38787, July 25, 2001]
|
|
|
Sec. 862.1215 Creatine phosphokinase/creatine kinase or isoenzymes test system.
|
|
(a) Identification. A creatine phosphokinase/creatine kinase or isoenzymes test system is a device intended to measure the activity of the enzyme creatine phosphokinase or its isoenzymes (a group of enzymes with similar biological activity) in plasma and serum. Measurements of creatine phosphokinase and its isoenzymes are used in the diagnosis and treatment of myocardial infarction and muscle diseases such as progressive, Duchenne-type muscular dystrophy.
(b) Classification. Class II.
|
|
|
Sec. 862.1220 Acute kidney injury test system.
|
|
(a) Identification. An acute kidney injury test system is a device that is intended to measure one or more analytes in human samples as an aid in the assessment of a patient's risk for developing acute kidney injury. Test results are intended to be used in conjunction with other clinical and diagnostic findings, consistent with professional standards of practice, including confirmation by alternative methods.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) Premarket notification submissions must detail an appropriate end user device training program that will be offered while marketing the device as part of your efforts to mitigate the risk of incorrect interpretation of test results.
(2) As part of the risk management activities performed as part of your 21 CFR 820.30 design controls, you must document the appropriate end user device training program provided in your premarket notification submission to satisfy special control 21 CFR 862.1220(b)(1) that will be offered while marketing the device as part of your efforts to mitigate the risk of incorrect interpretation of test results.
(3) Robust clinical data demonstrating the positive predictive value, negative predictive value, sensitivity and specificity of the test in the intended use population must be submitted as part of the premarket notification submission.
[82 FR 50072, Oct. 30, 2017]
|
|
|
Sec. 862.1225 Creatinine test system.
|
|
(a) Identification. A creatinine test system is a device intended to measure creatinine levels in plasma and urine. Creatinine measurements are used in the diagnosis and treatment of renal diseases, in monitoring renal dialysis, and as a calculation basis for measuring other urine analytes.
(b) Classification. Class II.
|
|
|
Sec. 862.1230 Cyclic AMP test system.
|
|
(a) Identification. A cyclic AMP test system is a device intended to measure the level of adenosine 3', 5'-monophosphate (cyclic AMP) in plasma, urine, and other body fluids. Cyclic AMP measurements are used in the diagnosis and treatment of endocrine disorders, including hyperparathyroidism (overactivity of the parathyroid gland). Cyclic AMP measurements may also be used in the diagnosis and treatment of Graves' disease (a disorder of the thyroid) and in the differentiation of causes of hypercalcemia (elevated levels of serum calcium.)
(b) Classification. Class II.
|
|
|
Sec. 862.1235 Cyclosporine test system.
|
|
(a) Identification. A cyclosporine test system is a device intended to quantitatively determine cyclosporine concentrations as an aid in the management of transplant patients receiving therapy with this drug. This generic type of device includes immunoassays and chromatographic assays for cyclosporine.
(b) Classification. Class II (special controls). The special control is "Class II Special Controls Guidance Document: Cyclosporine and Tacrolimus Assays; Guidance for Industry and FDA." See § 862.1(d) for the availability of this guidance document.
[67 FR 58329, Sept. 16, 2002]
|
|
|
Sec. 862.1240 Cystine test system.
|
|
(a) Identification. A cystine test system is a device intended to measure the amino acid cystine in urine. Cystine measurements are used in the diagnosis of cystinuria (occurrence of cystine in urine). Patients with cystinuria frequently develop kidney calculi (stones).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2305, Jan. 14, 2000]
|
|
|
Sec. 862.1245 Dehydroepiandrosterone (free and sulfate) test system.
|
|
(a) Identification. A dehydroepiandrosterone (free and sulfate) test system is a device intended to measure dehydroepiandrosterone (DHEA) and its sulfate in urine, serum, plasma, and amniotic fluid. Dehydroepiandrosterone measurements are used in the diagnosis and treatment of DHEA-secreting adrenal carcinomas.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1250 Desoxycorticosterone test system.
|
|
(a) Identification. A desoxycorticosterone test system is a device intended to measure desoxycorticosterone (DOC) in plasma and urine. DOC measurements are used in the diagnosis and treatment of patients with hypermineralocorticoidism (excess retention of sodium and loss of potassium) and other disorders of the adrenal gland.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1255 2,3-Diphosphoglyceric acid test system.
|
|
(a) Identification. A 2,3-diphosphoglyceric acid test system is a device intended to measure 2,3-diphosphoglyceric acid (2,3-DPG) in erythrocytes (red blood cells). Measurements of 2,3-diphosphoglyceric acid are used in the diagnosis and treatment of blood disorders that affect the delivery of oxygen by erythrocytes to tissues and in monitoring the quality of stored blood.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38787, July 25, 2001]
|
|
|
Sec. 862.1260 Estradiol test system.
|
|
(a) Identification. An estradiol test system is a device intended to measure estradiol, an estrogenic steroid, in plasma. Estradiol measurements are used in the diagnosis and treatment of various hormonal sexual disorders and in assessing placental function in complicated pregnancy.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1265 Estriol test system.
|
|
(a) Identification. An estriol test system is a device intended to measure estriol, an estrogenic steroid, in plasma, serum, and urine of pregnant females. Estriol measurements are used in the diagnosis and treatment of fetoplacental distress in certain cases of high-risk pregnancy.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1270 Estrogens (total, in pregnancy) test system.
|
|
(a) Identification. As estrogens (total, in pregnancy) test system is a device intended to measure total estrogens in plasma, serum, and urine during pregnancy. The device primarily measures estrone plus estradiol. Measurements of total estrogens are used to aid in the diagnosis and treatment of fetoplacental distress in certain cases of high-risk pregnancy.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1275 Estrogens (total, nonpregnancy) test system.
|
|
(a) Identification. As estrogens (total, nonpregnancy) test system is a device intended to measure the level of estrogens (total estrone, estradiol, and estriol) in plasma, serum, and urine of males and nonpregnant females. Measurement of estrogens (total, nonpregnancy) is used in the diagnosis and treatment of numerous disorders, including infertility, amenorrhea (absence of menses) differentiation of primary and secondary ovarian malfunction, estrogen secreting testicular and ovarian tumors, and precocious puberty in females.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1280 Estrone test system.
|
|
(a) Identification. An estrone test system is a device intended to measure estrone, an estrogenic steroid, in plasma. Estrone measurements are used in the diagnosis and treatment of numerous disorders, including infertility, amenorrhea, differentiation of primary and secondary ovarian malfunction, estrogen secreting testicular and ovarian tumors, and precocious puberty in females.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1285 Etiocholanolone test system.
|
|
(a) Identification. An etiocholanolone test system is a device intended to measure etiocholanolone in serum and urine. Etiocholanolone is a metabolic product of the hormone testosterone and is excreted in the urine. Etiocholanolone measurements are used in the diagnosis and treatment of disorders of the testes and ovaries.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1290 Fatty acids test system.
|
|
(a) Identification. A fatty acids test system is a device intended to measure fatty acids in plasma and serum. Measurements of fatty acids are used in the diagnosis and treatment of various disorders of lipid metabolism.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38787, July 25, 2001]
|
|
|
Sec. 862.1295 Folic acid test system.
|
|
(a) Identification. A folic acid test system is a device intended to measure the vitamin folic acid in plasma and serum. Folic acid measurements are used in the diagnosis and treatment of megaloblastic anemia, which is characterized by the presence of megaloblasts (an abnormal red blood cell series) in the bone marrow.
(b) Classification. Class II.
[52 FR 16122, May 1, 1987; 53 FR 11645, Apr. 8, 1988]
|
|
|
Sec. 862.1300 Follicle-stimulating hormone test system.
|
|
(a) Identification. A follicle-stimulating hormone test system is a device intended to measure follicle-stimulating hormone (FSH) in plasma, serum, and urine. FSH measurements are used in the diagnosis and treatment of pituitary gland and gonadal disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1305 Formiminoglutamic acid (FIGLU) test system.
|
|
(a) Identification. A formiminoglutamic acid (FIGLU) test system is a device intended to measure formiminolutamic acid in urine. FIGLU measurements obtained by this device are used in the diagnosis of anemias, such as pernicious anemia and congenital hemolytic anemia.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38787, July 25, 2001]
|
|
|
Sec. 862.1310 Galactose test system.
|
|
(a) Identification. A galactose test system is a device intended to measure galactose in blood and urine. Galactose measurements are used in the diagnosis and treatment of the hereditary disease galactosemia (a disorder of galactose metabolism) in infants.
(b) Classification. Class I.
|
|
|
Sec. 862.1315 Galactose-1-phosphate uridyl transferase test system.
|
|
(a) Identification. A galactose-1-phosphate uridyl transferase test system is a device intended to measure the activity of the enzyme galactose-1-phosphate uridyl transferase in erythrocytes (red blood cells). Measurements of galactose-1-phosphate uridyl transferase are used in the diagnosis and treatment of the hereditary disease galactosemia (disorder of galactose metabolism) in infants.
(b) Classification. Class II.
|
|
|
Sec. 862.1320 Gastric acidity test system.
|
|
(a) Identification. A gastric acidity test system is a device intended to measure the acidity of gastric fluid. Measurements of gastric acidity are used in the diagnosis and treatment of patients with peptic ulcer, Zollinger-Ellison syndrome (peptic ulcer due to gastrin-secreting tumor of the pancreas), and related gastric disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38787, July 25, 2001]
|
|
|
Sec. 862.1325 Gastrin test system.
|
|
(a) Identification. A gastrin test system is a device intended to measure the hormone gastrin in plasma and serum. Measurements of gastrin are used in the diagnosis and treatment of patients with ulcers, pernicious anemia, and the Zollinger-Ellison syndrome (peptic ulcer due to a gastrin-secreting tumor of the pancreas).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1330 Globulin test system.
|
|
(a) Identification. A globulin test system is a device intended to measure globulins (proteins) in plasma and serum. Measurements of globulin are used in the diagnosis and treatment of patients with numerous illnesses including severe liver and renal disease, multiple myeloma, and other disorders of blood globulins.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1335 Glucagon test system.
|
|
(a) Identification. A glucagon test system is a device intended to measure the pancreatic hormone glucagon in plasma and serum. Glucagon measurements are used in the diagnosis and treatment of patients with various disorders of carbohydrate metabolism, including diabetes mellitus, hypoglycemia, and hyperglycemia.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1340 Urinary glucose (nonquantitative) test system.
|
|
(a) Identification. A urinary glucose (nonquantitative) test system is a device intended to measure glucosuria (glucose in urine). Urinary glucose (nonquantitative) measurements are used in the diagnosis and treatment of carbohydrate metabolism disorders including diabetes mellitus, hypoglycemia, and hyperglycemia.
(b) Classification. Class II.
|
|
|
Sec. 862.1345 Glucose test system.
|
|
(a) Identification. A glucose test system is a device intended to measure glucose quantitatively in blood and other body fluids. Glucose measurements are used in the diagnosis and treatment of carbohydrate metabolism disorders including diabetes mellitus, neonatal hypoglycemia, and idiopathic hypoglycemia, and of pancreatic islet cell carcinoma.
(b) Classification. Class II (special controls). The device, when it is solely intended for use as a drink to test glucose tolerance, is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019; 85 FR 18445, Apr. 2, 2020]
|
|
|
Sec. 862.1350 Continuous glucose monitor secondary alarm system.
|
|
(a) Identification. A continuous glucose monitor (CGM) secondary alarm system is identified as a device intended to be used as a secondary alarm for a CGM to enable immediate awareness for potential clinical intervention to help assure patient safety.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9. The special controls for this device are:
(1) Devices being marketed must include appropriate measures to protect against unauthorized access to data and unauthorized modification of data.
(2) The labeling must prominently and conspicuously display a warning that states "Dosing decisions should not be made based on this device. The user should follow instructions on the continuous glucose monitoring system."
(3) The labeling for the device must include a statement that reads "This device is not intended to replace self-monitoring practices as advised by a physician."
[82 FR 13550, Mar. 14, 2017, as amended at 84 FR 71796, Dec. 30, 2019; 86 FR 20283, Apr. 19, 2021]
|
|
|
Sec. 862.1355 Integrated continuous glucose monitoring system.
|
|
(a) Identification. An integrated continuous glucose monitoring system (iCGM) is intended to automatically measure glucose in bodily fluids continuously or frequently for a specified period of time. iCGM systems are designed to reliably and securely transmit glucose measurement data to digitally connected devices, including automated insulin dosing systems, and are intended to be used alone or in conjunction with these digitally connected medical devices for the purpose of managing a disease or condition related to glycemic control.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) Design verification and validation must include the following:
(i) Robust clinical data demonstrating the accuracy of the device in the intended use population.
(ii) The clinical data must include a comparison between iCGM values and blood glucose values in specimens collected in parallel that are measured on an FDA-accepted laboratory-based glucose measurement method that is precise and accurate, and that is traceable to a higher order (e.g., an internationally recognized reference material and/or method).
(iii) The clinical data must be obtained from a clinical study designed to fully represent the performance of the device throughout the intended use population and throughout the measuring range of the device.
(iv) Clinical study results must demonstrate consistent analytical and clinical performance throughout the sensor wear period.
(v) Clinical study results in the adult population must meet the following performance requirements:
(A) For all iCGM measurements less than 70 milligrams/deciliter (mg/dL), the percentage of iCGM measurements within +/-15 mg/dL of the corresponding blood glucose value must be calculated, and the lower one-sided 95 percent confidence bound must exceed 85 percent.
(B) For all iCGM measurements from 70 mg/dL to 180 mg/dL, the percentage of iCGM measurements within +/-15 percent of the corresponding blood glucose value must be calculated, and the lower one-sided 95 percent confidence bound must exceed 70 percent.
(C) For all iCGM measurements greater than 180 mg/dL, the percentage of iCGM measurements within +/-15 percent of the corresponding blood glucose value must be calculated, and the lower one-sided 95 percent confidence bound must exceed 80 percent.
(D) For all iCGM measurements less than 70 mg/dL, the percentage of iCGM measurements within +/-40 mg/dL of the corresponding blood glucose value must be calculated, and the lower one-sided 95 percent confidence bound must exceed 98 percent.
(E) For all iCGM measurements from 70 mg/dL to 180 mg/dL, the percentage of iCGM measurements within +/-40 percent of the corresponding blood glucose value must be calculated, and the lower one-sided 95 percent confidence bound must exceed 99 percent.
(F) For all iCGM measurements greater than180 mg/dL, the percentage of iCGM measurements within +/-40 percent of the corresponding blood glucose value must be calculated, and the lower one-sided 95 percent confidence bound must exceed 99 percent.
(G) Throughout the device measuring range, the percentage of iCGM measurements within +/-20 percent of the corresponding blood glucose value must be calculated, and the lower one-sided 95 percent confidence bound must exceed 87 percent.
(H) When iCGM values are less than 70 mg/dL, no corresponding blood glucose value shall read above 180 mg/dL.
(I) When iCGM values are greater than 180 mg/dL, no corresponding blood glucose value shall read less than 70 mg/dL.
(J) There shall be no more than 1 percent of iCGM measurements that indicate a positive glucose rate of change greater than 1 mg/dL per minute (/min) when the corresponding true negative glucose rate of change is less than â??2 mg/dL/min as determined by the corresponding blood glucose measurements.
(K) There shall be no more than 1 percent of iCGM measurements that indicate a negative glucose rate of change less than â??1 mg/dL/min when the corresponding true positive glucose rate of change is greater than 2 mg/dL/min as determined by the corresponding blood glucose measurements.
(vi) Data demonstrating similar accuracy and rate of change performance of the iCGM in the pediatric population as compared to that in the adult population, or alternatively a clinical and/or technical justification for why pediatric data are not needed, must be provided and determined by FDA to be acceptable and appropriate.
(vii) Data must demonstrate that throughout the claimed sensor life, the device does not allow clinically significant gaps in sensor data availability that would prevent any digitally connected devices from achieving their intended use.
(2) Design verification and validation must include a detailed strategy to ensure secure and reliable means of iCGM data transmission to provide real-time glucose readings at clinically meaningful time intervals to devices intended to receive the iCGM glucose data.
(3) Design verification and validation must include adequate controls established during manufacturing and at product release to ensure the released product meets the performance specifications as defined in paragraphs (b)(1) and (b)(2) of this section.
(4) The device must demonstrate clinically acceptable performance in the presence of clinically relevant levels of potential interfering substances that are reasonably present in the intended use population, including but not limited to endogenous substances and metabolites, foods, dietary supplements, and medications.
(5) The device must include appropriate measures to ensure that disposable sensors cannot be used beyond its claimed sensor wear period.
(6) Design verification and validation must include results obtained through a usability study that demonstrates that the intended user can use the device safely and obtain the expected glucose measurement accuracy.
(7) The labeling required under § 809.10(b) of this chapter must include a separate description of the following sensor performance data observed in the clinical study performed in conformance with paragraph (b)(1) of this section for each intended use population, in addition to separate sensor performance data for each different iCGM insertion or use sites (e.g., abdomen, arm, buttock):
(i) A description of the accuracy in the following blood glucose concentration ranges: less than 54 mg/dL, 54 mg/dL to less than 70 mg/dL, 70 to 180 mg/dL, greater than 180 to 250 mg/dL, and greater than 250 mg/dL.
(ii) A description of the accuracy of positive and negative rate of change data.
(iii) A description of the frequency and duration of gaps in sensor data.
(iv) A description of the true, false, missed, and correct alert rates and a description of the available glucose concentration alert settings, if applicable.
(v) A description of the observed duration of iCGM life for the device.
[87 FR 9238, Feb. 18, 2022]
|
|
|
Sec. 862.1356 Interoperable automated glycemic controller.
|
|
(a) Identification. An interoperable automated glycemic controller is a device intended to automatically calculate drug doses based on inputs such as glucose and other relevant physiological parameters, and to command the delivery of such drug doses from a connected infusion pump. Interoperable automated glycemic controllers are designed to reliably and securely communicate with digitally connected devices to allow drug delivery commands to be sent, received, executed, and confirmed. Interoperable automated glycemic controllers are intended to be used in conjunction with digitally connected devices for the purpose of maintaining glycemic control.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) Design verification and validation must include:
(i) An appropriate, as determined by FDA, clinical implementation strategy, including data demonstrating appropriate, as determined by FDA, clinical performance of the device for its intended use, including all of its indications for use.
(A) The clinical data must be representative of the performance of the device in the intended use population and in clinically relevant use scenarios and sufficient to demonstrate appropriate, as determined by FDA, clinical performance of the device for its intended use, including all of its indications for use.
(B) For devices indicated for use with multiple therapeutic agents for the same therapeutic effect (e.g., more than one type of insulin), data demonstrating performance with each product or, alternatively, an appropriate, as determined by FDA, clinical justification for why such data are not needed.
(C) When determined to be necessary by FDA, the strategy must include postmarket data collection to confirm safe real-world use and monitor for rare adverse events.
(ii) Results obtained through a human factors study that demonstrates that an intended user can safely use the device for its intended use.
(iii) A detailed and appropriate, as determined by FDA, strategy to ensure secure and reliable means of data transmission with other intended connected devices.
(iv) Specifications that are appropriate, as determined by FDA, for connected devices that shall be eligible to provide input to (e.g., specification of glucose sensor performance) or accept commands from (e.g., specifications for drug infusion pump performance) the controller, and a detailed strategy for ensuring that connected devices meet these specifications.
(v) Specifications for devices responsible for hosting the controller, and a detailed and appropriate, as determined by FDA, strategy for ensuring that the specifications are met by the hosting devices.
(vi) Documentation demonstrating that appropriate, as determined by FDA, measures are in place (e.g., validated device design features) to ensure that safe therapy is maintained when communication with digitally connected devices is interrupted, lost, or re-established after an interruption. Validation testing results must demonstrate that critical events that occur during a loss of communications (e.g., commands, device malfunctions, occlusions, etc.) are handled and logged appropriately during and after the interruption to maintain patient safety.
(vii) A detailed plan and procedure for assigning postmarket responsibilities including adverse event reporting, complaint handling, and investigations with the manufacturers of devices that are digitally connected to the controller.
(2) Design verification and validation documentation must include appropriate design inputs and design outputs that are essential for the proper functioning of the device that have been documented and include the following:
(i) Risk control measures to address device system hazards;
(ii) Design decisions related to how the risk control measures impact essential performance; and
(iii) A traceability analysis demonstrating that all hazards are adequately controlled and that all controls have been validated in the final device design.
(3) The device shall include appropriate, as determined by FDA, and validated interface specifications for digitally connected devices. These interface specifications shall, at a minimum, provide for the following:
(i) Secure authentication (pairing) to connected devices;
(ii) Secure, accurate, and reliable means of data transmission between the controller and connected devices;
(iii) Sharing of necessary state information between the controller and any connected devices (e.g., battery level, reservoir level, sensor use life, pump status, error conditions);
(iv) Ensuring that the controller continues to operate safely when data is received in a manner outside the bounds of the parameters specified;
(v) A detailed process and procedures for sharing the controller's interface specification with connected devices and for validating the correct implementation of that protocol; and
(vi) A mechanism for updating the controller software, including any software that is required for operation of the controller in a manner that ensures its safety and performance.
(4) The device design must ensure that a record of critical events is stored and accessible for an adequate period to allow for auditing of communications between digitally connected devices, and to facilitate the sharing of pertinent information with the responsible parties for those connected devices. Critical events to be stored by the controller must, at a minimum, include:
(i) Commands issued by the controller, and associated confirmations the controller receives from digitally connected devices;
(ii) Malfunctions of the controller and malfunctions reported to the controller by digitally connected devices (e.g., infusion pump occlusion, glucose sensor shut down);
(iii) Alarms and alerts and associated acknowledgements from the controller as well as those reported to the controller by digitally connected devices; and
(iv) Connectivity events (e.g., establishment or loss of communications).
(5) The device must only receive glucose input from devices cleared under § 862.1355 (integrated continuous glucose monitoring system), unless FDA determines an alternate type of glucose input device is designed appropriately to allow the controller to meet the special controls contained within this section.
(6) The device must only command drug delivery from devices cleared under § 880.5730 of this chapter (alternate controller enabled infusion pump), unless FDA determines an alternate type of drug infusion pump device is designed appropriately to allow the controller to meet the special controls contained within this section.
(7) An appropriate, as determined by FDA, training plan must be established for users and healthcare providers to assure the safety and performance of the device when used. This may include, but not be limited to, training on device contraindications, situations in which the device should not be used, notable differences in device functionality or features compared to similar alternative therapies, and information to help prescribers identify suitable candidate patients, as applicable.
(8) The labeling required under § 809.10(b) of this chapter must include:
(i) A contraindication for use in pediatric populations except to the extent clinical performance data or other available information demonstrates that it can be safely used in pediatric populations in whole or in part.
(ii) A prominent statement identifying any populations for which use of this device has been determined to be unsafe.
(iii) A prominent statement identifying by name the therapeutic agents that are compatible with the controller, including their identity and concentration, as appropriate.
(iv) The identity of those digitally connected devices with which the controller can be used, including descriptions of the specific system configurations that can be used, per the detailed strategy submitted under paragraph (b)(1)(iii) of this section.
(v) A comprehensive description of representative clinical performance in the hands of the intended user, including information specific to use in the pediatric use population, as appropriate.
(vi) A comprehensive description of safety of the device, including, for example, the incidence of severe hypoglycemia, diabetic ketoacidosis, and other relevant adverse events observed in a study conducted to satisfy paragraph (b)(1)(i) of this section.
(vii) For wireless connection enabled devices, a description of the wireless quality of service required for proper use of the device.
(viii) For any controller with hardware components intended for multiple patient reuse, instructions for safely reprocessing the hardware components between uses.
[87 FR 14172, Mar. 14, 2022]
|
|
|
Sec. 862.1358 Insulin therapy adjustment device.
|
|
(a) Identification. An insulin therapy adjustment device is a device intended to incorporate biological inputs, including glucose measurement data from a continuous glucose monitor, to recommend insulin therapy adjustments as an aid in optimizing insulin therapy regimens for patients with diabetes mellitus.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) Design verification and validation must include the following:
(i) A complete description of the required data inputs, including timeframe over which data inputs must be collected and number of data points required for accurate recommendations;
(ii) A complete description of the types of device outputs and insulin therapy adjustment recommendations, including how the recommendations are generated;
(iii) Robust data demonstrating the clinical validity of the device outputs and insulin therapy recommendations;
(iv) A robust assessment of all input data specifications, including accuracy requirements for continuous glucose monitors and other devices generating data inputs, to ensure accurate and reliable therapy adjustment recommendations. This assessment must include adequate clinical justification for each specification;
(v) A detailed strategy to ensure secure and reliable means of data transmission to and from the device, including data integrity checks, accuracy checks, reliability checks, and security measures;
(vi) Robust data demonstrating that users can understand and appropriately interpret recommendations generated by the device; and
(vii) An appropriate mitigation strategy to minimize the occurrence of dosing recommendation errors, and to mitigate the risk to patients of any residual dosing recommendation errors to a clinically acceptable level.
(2) The device must not be intended for use in implementing automated insulin dosing.
(3) Your 21 CFR 809.10(b) labeling must include:
(i) The identification of specific insulin formulations that have been demonstrated to be compatible with use of the device;
(ii) A detailed description of the specifications of compatible devices that provide acceptable input data (e.g., continuous glucose monitors, insulin pumps) used to provide accurate and reliable therapy adjustment recommendations;
(iii) A detailed description of all types of required data (inputs) and dosing recommendations (outputs) that are provided by the device; and
(iv) A description of device limitations, and instructions to prevent possible disruption of accurate therapy adjustment recommendations (e.g., time zone changes due to travel).
[83 FR 54874, Nov. 1, 2018]
|
|
|
Sec. 862.1360 Gamma-glutamyl transpeptidase and isoenzymes test system.
|
|
(a) Identification. A gamma-glutamyl transpeptidase and isoenzymes test system is a device intended to measure the activity of the enzyme gamma-glutamyl transpeptidase (GGTP) in plasma and serum. Gamma-glutamyl transpeptidase and isoenzymes measurements are used in the diagnosis and treatment of liver diseases such as alcoholic cirrhosis and primary and secondary liver tumors.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1365 Glutathione test system.
|
|
(a) Identification. A glutathione test system is a device intended to measure glutathione (the tripeptide of glycine, cysteine, and glutamic acid) in erythrocytes (red blood cells). Glutathione measurements are used in the diagnosis and treatment of certain drug-induced hemolytic (erythrocyte destroying) anemias due to an inherited enzyme deficiency.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38787, July 25, 2001]
|
|
|
Sec. 862.1370 Human growth hormone test system.
|
|
(a) Identification. A human growth hormone test system is a device intended to measure the levels of human growth hormone in plasma. Human growth hormone measurements are used in the diagnosis and treatment of disorders involving the anterior lobe of the pituitary gland.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1373 Hemoglobin A1c test system.
|
|
(a) Identification. A hemoglobin A1c test system is a device used to measure the percentage concentration of hemoglobin A1c in blood. Measurement of hemoglobin A1c is used as an aid in the diagnosis of diabetes mellitus and as an aid in the identification of patients at risk for developing diabetes mellitus.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) The device must have initial and annual standardization verification by a certifying glycohemoglobin standardization organization deemed acceptable by FDA.
(2) The premarket notification submission must include performance testing to evaluate precision, accuracy, linearity, and interference, including the following:
(i) Performance testing of device precision must, at a minimum, use blood samples with concentrations near 5.0 percent, 6.5 percent, 8.0 percent, and 12 percent hemoglobin A1c. This testing must evaluate precision over a minimum of 20 days using at least three lots of the device and three instruments, as applicable.
(ii) Performance testing of device accuracy must include a minimum of 120 blood samples that span the measuring interval of the device and compare results of the new device to results of a standardized test method. Results must demonstrate little or no bias versus the standardized method.
(iii) Total error of the new device must be evaluated using single measurements by the new device compared to results of the standardized test method, and this evaluation must demonstrate a total error less than or equal to 6 percent.
(iv) Performance testing must demonstrate that there is little to no interference from common hemoglobin variants, including Hemoglobin C, Hemoglobin D, Hemoglobin E, Hemoglobin A2, and Hemoglobin S.
(3) When assay interference from Hemoglobin F or interference with other hemoglobin variants with low frequency in the population is observed, a warning statement must be placed in a black box and must appear in all labeling material for these devices describing the interference and any affected populations.
[79 FR 50551, Aug. 25, 2014]
|
|
|
Sec. 862.1375 Histidine test system.
|
|
(a) Identification. A histidine test system is a device intended to measure free histidine (an amino acid) in plasma and urine. Histidine measurements are used in the diagnosis and treatment of hereditary histidinemia characterized by excess histidine in the blood and urine often resulting in mental retardation and disordered speech development.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1377 Urinary homocystine (nonquantitative) test system.
|
|
(a) Identification. A urinary homocystine (nonquantitative) test system is a device intended to identify homocystine (an analogue of the amino acid cystine) in urine. The identification of urinary homocystine is used in the diagnosis and treatment of homocystinuria (homosystine in urine), a heritable metabolic disorder which may cause mental retardation.
(b) Classification. Class II.
|
|
|
Sec. 862.1380 Hydroxybutyric dehydrogenase test system.
|
|
(a) Identification. A hydroxybutyric dehydrogenase test system is a device intended to measure the activity of the enzyme alpha-hydroxybutric dehydrogenase (HBD) in plasma or serum. HBD measurements are used in the diagnosis and treatment of myocardial infarction, renal damage (such as rejection of transplants), certain hematological diseases (such as acute leukemias and megaloblastic anemias) and, to a lesser degree, liver disease.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38787, July 25, 2001]
|
|
|
Sec. 862.1385 17-Hydroxycorticosteroids (17-ketogenic steroids) test system.
|
|
(a) Identification. A 17-hydroxycorticosteroids (17-ketogenic steroids) test system is a device intended to measure corticosteroids that possess a dihydroxyacetone
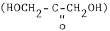
moiety on the steroid nucleus in urine. Corticosteroids with this chemical configuration include cortisol, cortisone 11-desoxycortisol, desoxycorticosterone, and their tetrahydroderivatives. This group of hormones is synthesized by the adrenal gland. Measurements of 17-hydroxycorticosteroids (17-ketogenic steroids) are used in the diagnosis and treatment of various diseases of the adrenal or pituitary glands and gonadal disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987; 52 FR 29468, Aug. 7, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1390 5-Hydroxyindole acetic acid/serotonin test system.
|
|
(a) Identification. A 5-hydroxyindole acetic acid/serotonin test system is a device intended to measure 5-hydroxyindole acetic acid/serotonin in urine. Measurements of 5-hydroxyindole acetic acid/serotonin are used in the diagnosis and treatment of carcinoid tumors of endocrine tissue.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1395 17-Hydroxyprogesterone test system.
|
|
(a) Identification. A 17-hydroxyprogesterone test system is a device intended to measure 17-hydroxyprogesterone (a steroid) in plasma and serum. Measurements of 17-hydroxyprogesterone are used in the diagnosis and treatment of various disorders of the adrenal glands or the ovaries.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1400 Hydroxyproline test system.
|
|
(a) Identification. A hydroxyproline test system is a device intended to measure the amino acid hydroxyproline in urine. Hydroxyproline measurements are used in the diagnosis and treatment of various collagen (connective tissue) diseases, bone disease such as Paget's disease, and endocrine disorders such as hyperparathyroidism and hyperthyroidism.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1405 Immunoreactive insulin test system.
|
|
(a) Identification. An immunoreactive insulin test system is a device intended to measure immunoreactive insulin in serum and plasma. Immunoreactive insulin measurements are used in the diagnosis and treatment of various carbohydrate metabolism disorders, including diabetes mellitus, and hypoglycemia.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2306, Jan. 14, 2000]
|
|
|
Sec. 862.1410 Iron (non-heme) test system.
|
|
(a) Identification. An iron (non-heme) test system is a device intended to measure iron (non-heme) in serum and plasma. Iron (non-heme) measurements are used in the diagnosis and treatment of diseases such as iron deficiency anemia, hemochromatosis (a disease associated with widespread deposit in the tissues of two iron-containing pigments, hemosiderin and hemofuscin, and characterized by pigmentation of the skin), and chronic renal disease.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1415 Iron-binding capacity test system.
|
|
(a) Identification. An iron-binding capacity test system is a device intended to measure iron-binding capacity in serum. Iron-binding capacity measurements are used in the diagnosis and treatment of anemia.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1420 Isocitric dehydrogenase test system.
|
|
(a) Identification. An isocitric dehydrogenase test system is a device intended to measure the activity of the enzyme isocitric dehydrogenase in serum and plasma. Isocitric dehydrogenase measurements are used in the diagnosis and treatment of liver disease such as viral hepatitis, cirrhosis, or acute inflammation of the biliary tract; pulmonary disease such as pulmonary infarction (local arrest or sudden insufficiency of the blood supply to the lungs), and diseases associated with pregnancy.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1430 17-Ketosteroids test system.
|
|
(a) Identification. A 17-ketosteroids test system is a device intended to measure 17-ketosteroids in urine. Measurements of 17-ketosteroids are used in the diagnosis and treatment of disorders of the adrenal cortex and gonads and of other endocrine disorders, including hypertension, diabetes, and hypothyroidism.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1435 Ketones (nonquantitative) test system.
|
|
(a) Identification. A ketones (nonquantitative) test system is a device intended to identify ketones in urine and other body fluids. Identification of ketones is used in the diagnosis and treatment of acidosis (a condition characterized by abnormally high acidity of body fluids) or ketosis (a condition characterized by increased production of ketone bodies such as acetone) and for monitoring patients on ketogenic diets and patients with diabetes.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1440 Lactate dehydrogenase test system.
|
|
(a) Identification. A lactate dehydrogenase test system is a device intended to measure the activity of the enzyme lactate dehydrogenase in serum. Lactate dehydrogenase measurements are used in the diagnosis and treatment of liver diseases such as acute viral hepatitis, cirrhosis, and metastatic carcinoma of the liver, cardiac diseases such as myocardial infarction, and tumors of the lung or kidneys.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 63 FR 59225, Nov. 3, 1998]
|
|
|
Sec. 862.1445 Lactate dehydrogenase isoenzymes test system.
|
|
(a) Identification. A lactate dehydrogenase isoenzymes test system is a device intended to measure the activity of lactate dehydrogenase isoenzymes (a group of enzymes with similar biological activity) in serum. Measurements of lactate dehydrogenase isoenzymes are used in the diagnosis and treatment of liver diseases, such as viral hepatitis, and myocardial infarction.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1450 Lactic acid test system.
|
|
(a) Identification. A lactic acid test system is a device intended to measure lactic acid in whole blood and plasma. Lactic acid measurements that evaluate the acid-base status are used in the diagnosis and treatment of lactic acidosis (abnormally high acidity of the blood).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1455 Lecithin/sphingomyelin ratio in amniotic fluid test system.
|
|
(a) Identification. A lecithin/sphingomyelin ratio in amniotic fluid test system is a device intended to measure the lecithin/sphingomyelin ratio in amniotic fluid. Lecithin and sphingomyelin are phospholipids (fats or fat-like substances containing phosphorus). Measurements of the lecithin/sphingomyelin ratio in amniotic fluid are used in evaluating fetal maturity.
(b) Classification. Class II.
|
|
|
Sec. 862.1460 Leucine aminopeptidase test system.
|
|
(a) Identification. A leucine aminopeptidase test system is a device intended to measure the activity of the enzyme leucine amino-peptidase in serum, plasma, and urine. Leucine aminopeptidase measurements are used in the diagnosis and treatment of liver diseases such as viral hepatitis and obstructive jaundice.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1465 Lipase test system.
|
|
(a) Identification. A lipase test system is a device intended to measure the activity of the enzymes lipase in serum. Lipase measurements are used in diagnosis and treatment of diseases of the pancreas such as acute pancreatitis and obstruction of the pancreatic duct.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1470 Lipid (total) test system.
|
|
(a) Identification. A lipid (total) test system is a device intended to measure total lipids (fats or fat-like substances) in serum and plasma. Lipid (total) measurements are used in the diagnosis and treatment of various diseases involving lipid metabolism and atherosclerosis.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1475 Lipoprotein test system.
|
|
(a) Identification. A lipoprotein test system is a device intended to measure lipoprotein in serum and plasma. Lipoprotein measurements are used in the diagnosis and treatment of lipid disorders (such as diabetes mellitus), atherosclerosis, and various liver and renal diseases.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1485 Luteinizing hormone test system.
|
|
(a) Identification. A luteinizing hormone test system is a device intended to measure luteinizing hormone in serum and urine. Luteinizing hormone measurements are used in the diagnosis and treatment of gonadal dysfunction.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1490 Lysozyme (muramidase) test system.
|
|
(a) Identification. A lysozyme (muramidase) test system is a device intended to measure the activity of the bacteriolytic enzyme lysozyme (muramidase) in serum, plasma, leukocytes, and urine. Lysozyme measurements are used in the diagnosis and treatment of monocytic leukemia and kidney disease.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1495 Magnesium test system.
|
|
(a) Identification. A magnesium test system is a device intended to measure magnesium levels in serum and plasma. Magnesium measurements are used in the diagnosis and treatment of hypomagnesemia (abnormally low plasma levels of magnesium) and hypermagnesemia (abnormally high plasma levels of magnesium).
(b) Classification. Class I.
|
|
|
Sec. 862.1500 Malic dehydrogenase test system.
|
|
(a) Identification. A malic dehydrogenase test system is a device that is intended to measure the activity of the enzyme malic dehydrogenase in serum and plasma. Malic dehydrogenase measurements are used in the diagnosis and treatment of muscle and liver diseases, myocardial infarctions, cancer, and blood disorders such as myelogenous (produced in the bone marrow) leukemia.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1505 Mucopolysaccharides (nonquantitative) test system.
|
|
(a) Identification. A mucopolysaccharides (nonquantitative) test system is a device intended to measure the levels of mucopolysaccharides in urine. Mucopolysaccharide measurements in urine are used in the diagnosis and treatment of various inheritable disorders that affect bone and connective tissues, such as Hurler's, Hunter's, Sanfilippo's, Scheie's Morquio's and Maroteaux-Lamy syndromes.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1509 Methylmalonic acid (nonquantitative) test system.
|
|
(a) Identification. A methylmalonic acid (nonquantitative) test system is a device intended to identify methylmalonic acid in urine. The identification of methylmalonic acid in urine is used in the diagnosis and treatment of methylmalonic aciduria, a heritable metabolic disorder which, if untreated, may cause mental retardation.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1510 Nitrite (nonquantitative) test system.
|
|
(a) Identification. A nitrite (nonquantitative) test system is a device intended to identify nitrite in urine. Nitrite identification is used in the diagnosis and treatment of uninary tract infection of bacterial origin.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1515 Nitrogen (amino-nitrogen) test system.
|
|
(a) Identification. A nitrogen (amino-nitrogen) test system is a device intended to measure amino acid nitrogen levels in serum, plasma, and urine. Nitrogen (amino-nitrogen) measurements are used in the diagnosis and treatment of certain forms of severe liver disease and renal disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1520 5'-Nucleotidase test system.
|
|
(a) Identification. A 5'-nucleotidase test system is a device intended to measure the activity of the enzyme 5'-nucleotidase in serum and plasma. Measurements of 5'-nucleotidase are used in the diagnosis and treatment of liver diseases and in the differentiations between liver and bone diseases in the presence of elevated serum alkaline phosphatase activity.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1530 Plasma oncometry test system.
|
|
(a) Identification. A plasma oncometry test system is a device intended to measure plasma oncotic pressure. Plasma oncotic pressure is that portion of the total fluid pressure contributed by proteins and other molecules too large to pass through a specified membrane. Measurements of plasma oncotic pressure are used in the diagnosis and treatment of dehydration and circulatory disorders related to low serum protein levels and increased capillary permeability, such as edema and shock.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1535 Ornithine carbamyl transferase test system.
|
|
(a) Identification. An ornithine carbamyl transferase test system is a device intended to measure the activity of the enzyme ornithine carbamyl transferase (OCT) in serum. Ornithine carbamyl transferase measurements are used in the diagnosis and treatment of liver diseases, such as infectious hepatitis, acute cholecystitis (inflammation of the gall bladder), cirrhosis, and liver metastases.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1540 Osmolality test system.
|
|
(a) Identification. An osmolality test system is a device intended to measure ionic and nonionic solute concentration in body fluids, such as serum and urine. Osmolality measurement is used as an adjunct to other tests in the evaluation of a variety of diseases, including kidney diseases (e.g., chronic progressive renal failure), diabetes insipidus, other endocrine and metabolic disorders, and fluid imbalances.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1542 Oxalate test system.
|
|
(a) Identification. An oxalate test system is a device intended to measure the concentration of oxalate in urine. Measurements of oxalate are used to aid in the diagnosis or treatment of urinary stones or certain other metabolic disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1545 Parathyroid hormone test system.
|
|
(a) Identification. A parathyroid hormone test system is a device intended to measure the levels of parathyroid hormone in serum and plasma. Measurements of parathyroid hormone levels are used in the differential diagnosis of hypercalcemia (abnormally high levels of calcium in the blood) and hypocalcemia (abnormally low levels of calcium in the blood) resulting from disorders of calcium metabolism.
(b) Classification. Class II.
|
|
|
Sec. 862.1550 Urinary pH (nonquantitative) test system.
|
|
(a) Identification. A urinary pH (nonquantitative) test system is a device intended to estimate the pH of urine. Estimations of pH are used to evaluate the acidity or alkalinity of urine as it relates to numerous renal and metabolic disorders and in the monitoring of patients with certain diets.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1555 Phenylalanine test system.
|
|
(a) Identification. A phenylalanine test system is a device intended to measure free phenylalanine (an amino acid) in serum, plasma, and urine. Measurements of phenylalanine are used in the diagnosis and treatment of congenital phenylketonuria which, if untreated, may cause mental retardation.
(b) Classification. Class II.
|
|
|
Sec. 862.1560 Urinary phenylketones (nonquantitative) test system.
|
|
(a) Identification. A urinary phenylketones (nonquantitative) test system is a device intended to identify phenylketones (such as phenylpyruvic acid) in urine. The identification of urinary phenylketones is used in the diagnosis and treatment of congenital phenylketonuria which, if untreated, may cause mental retardation.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1565 6-Phosphogluconate dehydrogenase test system.
|
|
(a) Identification. A 6-phosphogluconate dehydrogenase test system is a device intended to measure the activity of the enzyme 6-phosphogluconate dehydrogenase (6 PGD) in serum and erythrocytes. Measurements of 6-phosphogluconate dehydrogenase are used in the diagnosis and treatment of certain liver diseases (such as hepatitis) and anemias.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1570 Phosphohexose isomerase test system.
|
|
(a) Identification. A phosphohexose isomerase test system is a device intended to measure the activity of the enzyme phosphohexose isomerase in serum. Measurements of phosphohexose isomerase are used in the diagnosis and treatment of muscle diseases such as muscular dystrophy, liver diseases such as hepatitis or cirrhosis, and metastatic carcinoma.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1575 Phospholipid test system.
|
|
(a) Identification. A phospholipid test system is a device intended to measure phospholipids in serum and plasma. Measurements of phospholipids are used in the diagnosis and treatment of disorders involving lipid (fat) metabolism.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1580 Phosphorus (inorganic) test system.
|
|
(a) Identification. A phosphorus (inorganic) test system is a device intended to measure inorganic phosphorus in serum, plasma, and urine. Measurements of phosphorus (inorganic) are used in the diagnosis and treatment of various disorders, including parathyroid gland and kidney diseases, and vitamin D imbalance.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1585 Human placental lactogen test system.
|
|
(a) Identification. A human placental lactogen test system is a device intended to measure the hormone human placental lactogen (HPL), (also known as human chorionic somatomammotrophin (HCS)), in maternal serum and maternal plasma. Measurements of human placental lactogen are used in the diagnosis and clinical management of high-risk pregnancies involving fetal distress associated with placental insufficiency. Measurements of HPL are also used in pregnancies complicated by hypertension, proteinuria, edema, post-maturity, placental insufficiency, or possible miscarriage.
(b) Classification. Class II.
|
|
|
Sec. 862.1590 Porphobilinogen test system.
|
|
(a) Identification. A porphobilinogen test system is a device intended to measure porphobilinogen (one of the derivatives of hemoglobin which can make the urine a red color) in urine. Measurements obtained by this device are used in the diagnosis and treatment of porphyrias (primarily inherited diseases associated with disturbed porphyrine metabolism), lead poisoning, and other diseases characterized by alterations in the heme pathway.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2307, Jan. 14, 2000]
|
|
|
Sec. 862.1595 Porphyrins test system.
|
|
(a) Identification. A porphyrins test system is a device intended to measure porphyrins (compounds formed during the biosynthesis of heme, a constituent of hemoglobin, and related compounds) in urine and feces. Measurements obtained by this device are used in the diagnosis and treatment of lead poisoning, porphyrias (primarily inherited diseases associated with disturbed porphyrin metabolism), and other diseases characterized by alterations in the heme pathway.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1600 Potassium test system.
|
|
(a) Identification. A potassium test system is a device intended to measure potassium in serum, plasma, and urine. Measurements obtained by this device are used to monitor electrolyte balance in the diagnosis and treatment of diseases conditions characterized by low or high blood potassium levels.
(b) Classification. Class II.
|
|
|
Sec. 862.1605 Pregnanediol test system.
|
|
(a) Identification. A pregnanediol test system is a device intended to measure pregnanediol (a major urinary metabolic product of progesterone) in urine. Measurements obtained by this device are used in the diagnosis and treatment of disorders of the ovaries or placenta.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1610 Pregnanetriol test system.
|
|
(a) Identification. A pregnanetriol test system is a device intended to measure pregnanetriol (a precursor in the biosynthesis of the adrenal hormone cortisol) in urine. Measurements obtained by this device are used in the diagnosis and treatment of congenital adrenal hyperplasia (congenital enlargement of the adrenal gland).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1615 Pregnenolone test system.
|
|
(a) Identification. A pregnenolone test system is a device intended to measure pregnenolone (a precursor in the biosynthesis of the adrenal hormone cortisol and adrenal androgen) in serum and plasma. Measurements obtained by this device are used in the diagnosis and treatment of diseases of the adrenal cortex or the gonads.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1620 Progesterone test system.
|
|
(a) Identification. A progesterone test system is a device intended to measure progesterone (a female hormone) in serum and plasma. Measurements obtained by this device are used in the diagnosis and treatment of disorders of the ovaries or placenta.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1622 Prognostic test for assessment of liver related disease progression.
|
|
(a) Identification. A prognostic test for assessment of liver related disease progression is intended to measure one or more analytes obtained from human samples as an aid in assessing progression of liver related disease. This device is not intended for diagnosis of any disease, for monitoring the effect of any therapeutic product, for assessing progression to hepatocellular carcinoma, or for assessing disease progression in individuals with viral hepatitis. It is also not intended for the detection of viruses, viral antigens, or antibodies to viruses.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) Design verification and validation must include clinical validation data providing:
(i) Information demonstrating clinical performance in a population of patients with liver disease for the different risk categories (e.g., at lower risk, at higher risk) for progression of their disease using well characterized clinical specimens representing the intended use population collected from multiple intended clinical sites, or an alternative study design determined to be appropriate by FDA.
(ii) Information demonstrating that the outcomes measured and the length of followup are clinically relevant for the progression of the specified liver disease.
(iii) Information demonstrating that the clinical criteria for determining whether the target disease is present and that the exclusion and inclusion criteria for subjects who have the target disease are appropriate.
(iv) Information demonstrating test performance of the complete test system, including any sample collection and processing steps.
(v) Information, provided or referenced, generated in samples from non-diseased individuals, that demonstrate the upper and lower reference intervals for the output provided by the device.
(2) The labeling required under 21 CFR 809.10(b) must include:
(i) A warning statement that test results are not intended to diagnose disease or for monitoring the effect of any therapeutic product.
(ii) A warning statement that test results are intended to be used in conjunction with other clinical and diagnostic findings, consistent with professional standards of practice, including information obtained by alternative methods, and clinical evaluation, as appropriate.
(iii) A warning statement that describes any limitations on the clinical interpretation(s) of the test results.
(iv) Detailed information on device performance, including any limitations to the data generated in the clinical study(ies) and information on device performance in relevant subgroups (e.g., severity of liver disease at the beginning of the observation period) observed in the clinical study(ies).
(v) Information on the analytical performance of the device, including demonstration of reproducibility across multiple sites and multiple reagent lots, or an alternative reproducibility study design determined to be appropriate by FDA.
[88 FR 2599, Jan. 17, 2023]
|
|
|
Sec. 862.1625 Prolactin (lactogen) test system.
|
|
(a) Identification. A prolactin (lactogen) test system is a device intended to measure the anterior pituitary polypeptide hormone prolactin in serum and plasma. Measurements obtained by this device are used in the diagnosis and treatment of disorders of the anterior pituitary gland or of the hypothalamus portion of the brain.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1630 Protein (fractionation) test system.
|
|
(a) Identification. A protein (fractionation) test system is a device intended to measure protein fractions in blood, urine, cerebrospinal fluid, and other body fluids. Protein fractionations are used as an aid in recognizing abnormal proteins in body fluids and genetic variants of proteins produced in diseases with tissue destruction.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1635 Total protein test system.
|
|
(a) Identification. A total protein test system is a device intended to measure total protein(s) in serum or plasma. Measurements obtained by this device are used in the diagnosis and treatment of a variety of diseases involving the liver, kidney, or bone marrow as well as other metabolic or nutritional disorders.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 63 FR 59225, Nov. 3, 1998]
|
|
|
Sec. 862.1640 Protein-bound iodine test system.
|
|
(a) Identification. A protein-bound iodine test system is a device intended to measure protein-bound iodine in serum. Measurements of protein-bound iodine obtained by this device are used in the diagnosis and treatment of thyroid disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1645 Urinary protein or albumin (nonquantitative) test system.
|
|
(a) Identification. A urinary protein or albumin (nonquantitative) test system is a device intended to identify proteins or albumin in urine. Identification of urinary protein or albumin (nonquantitative) is used in the diagnosis and treatment of disease conditions such as renal or heart diseases or thyroid disorders, which are characterized by proteinuria or albuminuria.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1650 Pyruvate kinase test system.
|
|
(a) Identification. A pyruvate kinase test system is a device intended to measure the activity of the enzyme pyruvate kinase in erythrocytes (red blood cells). Measurements obtained by this device are used in the diagnosis and treatment of various inherited anemias due to pyruvate kinase deficiency or of acute leukemias.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1655 Pyruvic acid test system.
|
|
(a) Identification. A pyruvic acid test system is a device intended to measure pyruvic acid (an intermediate compound in the metabolism of carbohydrate) in plasma. Measurements obtained by this device are used in the evaluation of electrolyte metabolism and in the diagnosis and treatment of acid-base and electrolyte disturbances or anoxia (the reduction of oxygen in body tissues).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1660 Quality control material (assayed and unassayed).
|
|
(a) Identification. A quality control material (assayed and unassayed) for clinical chemistry is a device intended for medical purposes for use in a test system to estimate test precision and to detect systematic analytical deviations that may arise from reagent or analytical instrument variation. A quality control material (assayed and unassayed) may be used for proficiency testing in interlaboratory surveys. This generic type of device includes controls (assayed and unassayed) for blood gases, electrolytes, enzymes, multianalytes (all kinds), single (specified) analytes, or urinalysis controls.
(b) Classification. Class I (general controls). Except when intended for use in donor screening tests, quality control materials (assayed and unassayed) are exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000, 84 FR 71796, Dec. 30, 2019]
|
|
|
Sec. 862.1665 Sodium test system.
|
|
(a) Identification. A sodium test system is a device intended to measure sodium in serum, plasma, and urine. Measurements obtained by this device are used in the diagnosis and treatment of aldosteronism (excessive secretion of the hormone aldosterone), diabetes insipidus (chronic excretion of large amounts of dilute urine, accompanied by extreme thirst), adrenal hypertension, Addison's disease (caused by destruction of the adrenal glands), dehydration, inappropriate antidiuretic hormone secretion, or other diseases involving electrolyte imbalance.
(b) Classification. Class II.
|
|
|
Sec. 862.1670 Sorbitol dehydrogenase test system.
|
|
(a) Identification. A sorbitol dehydrogenase test system is a device intended to measure the activity of the enzyme sorbitol dehydrogenase in serum. Measurements obtained by this device are used in the diagnosis and treatment of liver disorders such as cirrhosis or acute hepatitis.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1675 Blood specimen collection device.
|
|
(a) Identification. A blood specimen collection device is a device intended for medical purposes to collect and to handle blood specimens and to separate serum from nonserum (cellular) components prior to further testing. This generic type device may include blood collection tubes, vials, systems, serum separators, blood collection trays, or vacuum sample tubes.
(b) Classification. Class II.
|
|
|
Sec. 862.1678 Tacrolimus test system.
|
|
(a) Identification. A tacrolimus test system is a device intended to quantitatively determine tacrolimus concentrations as an aid in the management of transplant patients receiving therapy with this drug. This generic type of device includes immunoassays and chromatographic assays for tacrolimus.
(b) Classification. Class II (special controls). The special control is "Class II Special Controls Guidance Document: Cyclosporine and Tacrolimus Assays; Guidance for Industry and FDA." See § 862.1(d) for the availability of this guidance document.
[67 FR 58329, Sept. 16, 2002]
|
|
|
Sec. 862.1680 Testosterone test system.
|
|
(a) Identification. A testosterone test system is a device intended to measure testosterone (a male sex hormone) in serum, plasma, and urine. Measurement of testosterone are used in the diagnosis and treatment of disorders involving the male sex hormones (androgens), including primary and secondary hypogonadism, delayed or precocious puberty, impotence in males and, in females hirsutism (excessive hair) and virilization (masculinization) due to tumors, polycystic ovaries, and adrenogenital syndromes.
(b) Classification. Class I.
[52 FR 16122, May 1, 1987; 53 FR 11645, Apr. 8, 1988]
|
|
|
Sec. 862.1685 Thyroxine-binding globulin test system.
|
|
(a) Identification. A thyroxine-binding globulin test system is a device intended to measure thyroxine (thyroid)-binding globulin (TBG), a plasma protein which binds thyroxine, in serum and plasma. Measurements obtained by this device are used in the diagnosis and treatment of thyroid diseases.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71797, Dec. 30, 2019]
|
|
|
Sec. 862.1690 Thyroid stimulating hormone test system.
|
|
(a) Identification. A thyroid stimulating hormone test system is a device intended to measure thyroid stimulating hormone, also known as thyrotrophin and thyrotrophic hormone, in serum and plasma. Measurements of thyroid stimulating hormone produced by the anterior pituitary are used in the diagnosis of thyroid or pituitary disorders.
(b) Classification. Class II.
|
|
|
Sec. 862.1695 Free thyroxine test system.
|
|
(a) Identification. A free thyroxine test system is a device intended to measure free (not protein bound) thyroxine (thyroid hormone) in serum or plasma. Levels of free thyroxine in plasma are thought to reflect the amount of thyroxine hormone available to the cells and may therefore determine the clinical metabolic status of thyroxine. Measurements obtained by this device are used in the diagnosis and treatment of thyroid diseases.
(b) Classification. Class II.
|
|
|
Sec. 862.1700 Total thyroxine test system.
|
|
(a) Identification. A total thyroxine test system is a device intended to measure total (free and protein bound) thyroxine (thyroid hormone) in serum and plasma. Measurements obtained by this device are used in the diagnosis and treatment of thyroid diseases.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71797, Dec. 30, 2019]
|
|
|
Sec. 862.1705 Triglyceride test system.
|
|
(a) Identification. A triglyceride test system is a device intended to measure triglyceride (neutral fat) in serum and plasma. Measurements obtained by this device are used in the diagnosis and treatment of patients with diabetes mellitus, nephrosis, liver obstruction, other diseases involving lipid metabolism, or various endocrine disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1710 Total triiodothyronine test system.
|
|
(a) Identification. A total triiodothyronine test system is a device intended to measure the hormone triiodothyronine in serum and plasma. Measurements obtained by this device are used in the diagnosis and treatment of thyroid diseases such as hyperthyroidism.
(b) Classification. Class II. This device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 62286, Oct. 18, 2000]
|
|
|
Sec. 862.1715 Triiodothyronine uptake test system.
|
|
(a) Identification. A triiodothyronine uptake test system is a device intended to measure the total amount of binding sites available for binding thyroid hormone on the thyroxine-binding proteins, thyroid-binding globulin, thyroxine-binding prealbumin, and albumin of serum and plasma. The device provides an indirect measurement of thyrkoxine levels in serum and plasma. Measurements of triiodothyronine uptake are used in the diagnosis and treatment of thyroid disorders.
(b) Classification. Class II. The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 64 FR 1124, Jan. 8, 1999]
|
|
|
Sec. 862.1720 Triose phosphate isomerase test system.
|
|
(a) Identification. A triose phosphate isomerase test system is a device intended to measure the activity of the enzyme triose phosphate isomerase in erythrocytes (red blood cells). Triose phosphate isomerase is an enzyme important in glycolysis (the energy-yielding conversion of glucose to lactic acid in various tissues). Measurements obtained by this device are used in the diagnosis and treatment of congenital triose phosphate isomerase enzyme deficiency, which causes a type of hemolytic anemia.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1725 Trypsin test system.
|
|
(a) Identification. A trypsin test system is a device intended to measure the activity of trypsin (a pancreatic enzyme important in digestion for the breakdown of proteins) in blood and other body fluids and in feces. Measurements obtained by this device are used in the diagnosis and treatment of pancreatic disease.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1730 Free tyrosine test system.
|
|
(a) Identification. A free tyrosine test system is a device intended to measure free tyrosine (an amono acid) in serum and urine. Measurements obtained by this device are used in the diagnosis and treatment of diseases such as congenital tyrosinemia (a disease that can cause liver/kidney disorders) and as an adjunct to the measurement of phenylalanine in detecting congenital phenylketonuria (a disease that can cause brain damage).
(b) Classification. Class I.
|
|
|
Sec. 862.1770 Urea nitrogen test system.
|
|
(a) Identification. A urea nitrogen test system is a device intended to measure urea nitrogen (an end-product of nitrogen metabolism) in whole blood, serum, plasma, and urine. Measurements obtained by this device are used in the diagnosis and treatment of certain renal and metabolic diseases.
(b) Classification. Class II.
|
|
|
Sec. 862.1775 Uric acid test system.
|
|
(a) Identification. A uric acid test system is a device intended to measure uric acid in serum, plasma, and urine. Measurements obtained by this device are used in the diagnosis and treatment of numerous renal and metabolic disorders, including renal failure, gout, leukemia, psoriasis, starvation or other wasting conditions, and of patients receiving cytotoxic drugs.
(b) Classification. Class I (general controls). The device, when it is solely intended for use as an acid reduction of ferric ion test, a phosphotungstate reduction test, a gasometric uricase test, an ultraviolet uricase test, or an oxygen rate uricase test, is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 84 FR 71797, Dec. 30, 2019; 85 FR 18445, Apr. 2, 2020]
|
|
|
Sec. 862.1780 Urinary calculi (stones) test system.
|
|
(a) Identification. A urinary calculi (stones) test system is a device intended for the analysis of urinary calculi. Analysis of urinary calculi is used in the diagnosis and treatment of calculi of the urinary tract.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1785 Urinary urobilinogen (nonquantitative) test system.
|
|
(a) Identification. A urinary urobilinogen (nonquantitative) test system is a device intended to detect and estimate urobilinogen (a bile pigment degradation product of red cell hemoglobin) in urine. Estimations obtained by this device are used in the diagnosis and treatment of liver diseases and hemolytic (red cells) disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1790 Uroporphyrin test system.
|
|
(a) Identification. A uroporphyrin test system is a device intended to measure uroporphyrin in urine. Measurements obtained by this device are used in the diagnosis and treatment of porphyrias (primarily inherited diseases associated with disturbed porphyrin metabolism), lead poisoning, and other diseases characterized by alterations in the heme pathway.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1795 Vanilmandelic acid test system.
|
|
(a) Identification. A vanilmandelic acid test system is a device intended to measure vanilmandelic acid in urine. Measurements of vanilmandelic acid obtained by this device are used in the diagnosis and treatment of neuroblastoma, pheochromocytoma, and certain hypertensive conditions.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1805 Vitamin A test system.
|
|
(a) Identification. A vitamin A test system is a device intended to measure vitamin A in serum or plasma. Measurements obtained by this device are used in the diagnosis and treatment of vitamin A deficiency conditions, including night blindness, or skin, eye, or intestinal disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1810 Vitamin B12 test system.
|
|
(a) Identification. A vitamin B12 test system is a device intended to measure vitamin B12 in serum, plasma, and urine. Measurements obtained by this device are used in the diagnosis and treatment of anemias of gastrointestinal malabsorption.
(b) Classification. Class II.
|
|
|
Sec. 862.1815 Vitamin E test system.
|
|
(a) Identification. A vitamin E test system is a device intended to measure vitamin E (tocopherol) in serum. Measurements obtained by this device are used in the diagnosis and treatment of infants with vitamin E deficiency syndrome.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 subject to the limitations in § 862.9.
[52 FR 16122, May 1, 1987, as amended at 53 FR 21449, June 8, 1988; 66 FR 38788, July 25, 2001]
|
|
|
Sec. 862.1820 Xylose test system.
|
|
(a) Identification. A xylose test system is a device intended to measure xylose (a sugar) in serum, plasma, and urine. Measurements obtained by this device are used in the diagnosis and treatment of gastrointestinal malabsorption syndrome (a group of disorders in which there is subnormal absorption of dietary constituents and thus excessive loss from the body of the nonabsorbed substances).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 862.9.
[52 FR 16122, May 1, 1987, as amended at 65 FR 2308, Jan. 14, 2000]
|
|
|
Sec. 862.1825 Vitamin D test system.
|
|
(a) Identification. A vitamin D test system is a device intended for use in clinical laboratories for the quantitative determination of 25-hydroxyvitamin D (25-OH-D) and other hydroxylated metabolites of vitamin D in serum or plasma to be used in the assessment of vitamin D sufficiency.
(b) Classification. Class II (special controls). Vitamin D test systems must comply with the following special controls:
(1) Labeling in conformance with 21 CFR 809.10 and
(2) Compliance with existing standards of the National Committee on Clinical Laboratory Standards.
[63 FR 40366, July 29, 1998]
|
|
|
Sec. 862.1840 Total 25-hydroxyvitamin D mass spectrometry test system.
|
|
(a) Identification. A total 25-hydroxyvitamin D mass spectrometry test system is a device intended for use in clinical laboratories for the quantitative determination of total 25-hydroxyvitamin D (25-OH-D) in serum or plasma to be used in the assessment of vitamin D sufficiency.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in part 807, subpart E, of this chapter subject to the limitations in § 862.9. The device must comply with the following special controls:
(1) The device must have initial and annual standardization verification by a certifying vitamin D standardization organization deemed acceptable by FDA.
(2) The 21 CFR 809.10(b) compliant labeling must include detailed descriptions of performance testing conducted to evaluate precision, accuracy, linearity, interference, including the following:
(i) Performance testing of device precision must, at a minimum, use intended sample type with Vitamin D concentrations at medically relevant decision points. At least one sample in the precision studies must be an unmodified patient sample. This testing must evaluate repeatability and reproducibility using a protocol from an FDA-recognized standard.
(ii) Performance testing of device accuracy must include a minimum of 115 serum or plasma samples that span the measuring interval of the device and compare results of the new device to results of a reference method or a legally marketed standardized mass spectrometry based vitamin D assay. The results must be described in the 21 CFR 809.10(b)(12) compliant labeling of the device.
(iii) Interference from vitamin D analogs and metabolites including vitamin D2, vitamin D3, 1-hydroxyvitamin D2, 1-hydroxyvitamin D3, 3-Epi-25-Hydroxyvitamin D2, 3-Epi-25-Hydroxyvitamin D3, 1,25-Dihydroxyvitamin D2, 1,25-Dihydroxyvitamin D3, 3-Epi-1,25-Dihydroxyvitamin D2, and 3-Epi-1,25-Dihydroxyvitamin D3, 25, 26-Dihydroxyvitamin-D3, 24 (R), 25-dihydroxyvitamin-D3, 23 (R), 25-dihydroxyvitamin-D3 must be described in the 21 CFR 809.10(b)(7) compliant labeling of the device.
(3) The 21 CFR 809.10(b) compliant labeling must be supported by a reference range study representative of the performance of the device. The study must be conducted using samples collected from apparently healthy male and female adults at least 21 years of age and older from at least 3 distinct climatic regions within the United States in different weather seasons. The ethnic, racial, and gender background of this study population must be representative of the U.S. population demographics.
(4) The results of the device as provided in the 21 CFR 809.10(b) compliant labeling and any test report generated must be reported as only total 25-hydroxyvitamin D.
[82 FR 51559, Nov. 7, 2017, as amended at 83 FR 25914, June 5, 2018]
|
|
|
Authority: 21 U.S.C. 351, 360, 360c, 360e, 360j, 360l, 371.
Source: 52 FR 16122, May 1, 1987, unless otherwise noted.
|
|





