|
|
| [Code of Federal Regulations] |
| [Title 21, Volume 8] |
| [CITE: 21CFR866] |
TITLE 21--FOOD AND DRUGS CHAPTER I--FOOD AND DRUG ADMINISTRATION DEPARTMENT OF HEALTH AND HUMAN SERVICES
SUBCHAPTER H - MEDICAL DEVICES
| |
| PART 866 | IMMUNOLOGY AND MICROBIOLOGY DEVICES |
|
|
|
|
Subpart F - Immunological Test Systems
|
|
Sec. 866.5040 Albumin immunological test system.
|
|
(a) Identification. An albumin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the albumin (a plasma protein) in serum and other body fluids. Measurement of albumin aids in the diagnosis of kidney and intestinal diseases.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 63 FR 59227, Nov. 3, 1998]
|
|
|
Sec. 866.5060 Prealbumin immunological test system.
|
|
(a) Identification. A prealbumin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the prealbumin (a plasma protein) in serum and other body fluids. Measurement of prealbumin levels in serum may aid in the assessment of the patient's nutritional status.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5065 Human allotypic marker immunological test system.
|
|
(a) Identification. A human allotypic marker immunological test system is a device that consists of the reagents used to identify by immunochemical techniques the inherited human protein allotypic markers (such as nGm, nA2 m, and Km allotypes) in serum and other body fluids. The identification may be used while studying population genetics.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5080 Alpha-1-antichymotrypsin immunological test system.
|
|
(a) Identification. An alpha -1-antichymotrypsin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques alpha -1-antichymotrypsin (a protein) in serum, other body fluids, and tissues. Alpha -1-antichymotrypsin helps protect tissues against proteolytic (protein-splitting) enzymes released during infection.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5090 Antimitochondrial antibody immunological test system.
|
|
(a) Identification. An antimitochondrial antibody immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the antimitochondrial antibodies in human serum. The measurements aid in the diagnosis of diseases that produce a spectrum of autoantibodies (antibodies produced against the body's own tissue), such as primary biliary cirrhosis (degeneration of liver tissue) and chronic active hepatitis (inflammation of the liver).
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5100 Antinuclear antibody immunological test system.
|
|
(a) Identification. An antinuclear antibody immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the autoimmune antibodies in serum, other body fluids, and tissues that react with cellular nuclear constituents (molecules present in the nucleus of a cell, such as ribonucleic acid, deoxyribonucleic acid, or nuclear proteins). The measurements aid in the diagnosis of systemic lupus erythematosus (a multisystem autoimmune disease in which antibodies attack the victim's own tissues), hepatitis (a liver disease), rheumatoid arthritis, Sjogren's syndrome (arthritis with inflammation of the eye, eyelid, and salivary glands), and systemic sclerosis (chronic hardening and shrinking of many body tissues).
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5110 Antiparietal antibody immunological test system.
|
|
(a) Identification. An antiparietal antibody immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the specific antibody for gastric parietal cells in serum and other body fluids. Gastric parietal cells are those cells located in the stomach that produce a protein that enables vitamin B12 to be absorbed by the body. The measurements aid in the diagnosis of vitamin B12 deficiency (or pernicious anemia), atrophic gastritis (inflammation of the stomach), and autoimmune connective tissue diseases (diseases resulting when the body produces antibodies against its own tissues).
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5120 Antismooth muscle antibody immunological test system.
|
|
(a) Identification. An antismooth muscle antibody immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the antismooth muscle antibodies (antibodies to nonstriated, involuntary muscle) in serum. The measurements aid in the diagnosis of chronic hepatitis (inflammation of the liver) and autoimmune connective tissue diseases (diseases resulting from antibodies produced against the body's own tissues).
(b) Classification Class II (performance standards).
|
|
|
Sec. 866.5130 Alpha-1-antitrypsin immunological test system.
|
|
(a) Identification. An alpha -1-antitrypsin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the alpha -1-antitrypsin (a plasma protein) in serum, other body fluids, and tissues. The measurements aid in the diagnosis of several conditions including juvenile and adult cirrhosis of the liver. In addition, alpha -1-antitrypsin deficiency has been associated with pulmonary emphysema.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5150 Bence-Jones proteins immunological test system.
|
|
(a) Identification. A Bence-Jones proteins immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the Bence-Jones proteins in urine and plasma. Immunoglobulin molecules normally consist of pairs of polypeptide chains (subunits) of unequal size (light chains and heavy chains) bound together by several disulfide bridges. In some cancerous conditions, there is a proliferation of one plasma cell (antibody-producing cell) with excess production of light chains of one specific kind (monoclonal light chains). These free homogeneous light chains not associated with an immunoglobulin molecule can be found in urine and plasma, and have been called Bence-Jones proteins. Measurement of Bence-Jones proteins and determination that they are monoclonal aid in the diagnosis of multiple myeloma (malignant proliferation of plasma cells), Waldenstrom's macroglobulinemia (increased production of large immunoglobulins by spleen and bone marrow cells), leukemia (cancer of the blood-forming organs), and lymphoma (cancer of the lymphoid tissue).
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5160 Beta-globulin immunological test system.
|
|
(a) Identification. A beta -globulin immunological test system is a device that consists of reagents used to measure by immunochemical techniques beta globulins (serum protein) in serum and other body fluids. Beta -globulin proteins include beta -lipoprotein, transferrin, glycoproteins, and complement, and are rarely associated with specific pathologic disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5170 Breast milk immunological test system.
|
|
(a) Identification. A breast milk immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the breast milk proteins.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 59 FR 63007, Dec. 7, 1994; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5180 Fecal calprotectin immunological test system.
|
|
(a) Identification. A fecal calprotectin immunological test system is an in vitro diagnostic device that consists of reagents used to quantitatively measure, by immunochemical techniques, fecal calprotectin in human stool specimens. The device is intended forin vitro diagnostic use as an aid in the diagnosis of inflammatory bowel diseases (IBD), specifically Crohn's disease and ulcerative colitis, and as an aid in differentiation of IBD from irritable bowel syndrome.
(b) Classification. Class II (special controls). The special control for these devices is FDA's guidance document entitled "Class II Special Controls Guidance Document: Fecal Calprotectin Immunological Test Systems." For the availability of this guidance document, see § 866.1(e).
[71 FR 42598, July 27, 2006]
|
|
|
Sec. 866.5200 Carbonic anhydrase B and C immunological test system.
|
|
(a) Identification. A carbonic anhydrase B and C immunological test system is a device that consists of the reagents used to measure by immunochemical techniques specific carbonic anhydrase protein molecules in serum and other body fluids. Measurements of carbonic anhydrase B and C aid in the diagnosis of abnormal hemoglobin metabolism.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5210 Ceruloplasmin immunological test system.
|
|
(a) Identification. A ceruloplasmin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the ceruloplasmin (copper-transporting serum protein) in serum, other body fluids, or tissues. Measurements of ceruloplasmin aid in the diagnosis of copper metabolism disorders.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 84 FR 71800, Dec. 30, 2019]
|
|
|
Sec. 866.5220 Cohn fraction II immunological test system.
|
|
(a) Identification. A Cohn fraction II immunological test system is a device that consists of the reagents that contain or are used to measure that fraction of plasma containing protein gamma globulins, predominantly of the IgG class. The device may be used as a coprecipitant in radioimmunoassay methods, as raw material for the purification of IgG subclasses, and to reduce nonspecific adsorption of plasma proteins in immunoassay techniques. Measurement of these proteins aids in the diagnosis of any disease concerned with abnormal levels of IgG gamma globulins such as agammaglobulinemia or multiple myeloma.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 59 FR 63007, Dec. 7, 1994; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5230 Colostrum immunological test system.
|
|
(a) Identification. A colostrum immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the specific proteins in colostrum. Colostrum is a substance excreted by the mammary glands during pregnancy and until production of breast milk begins 1 to 5 days after childbirth.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 59 FR 63007, Dec. 7, 1994; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5240 Complement components immunological test system.
|
|
(a) Identification. A complement components immunological test system is a device that consists of the reagents used to measure by immunochemical techniques complement components C1q, C1r, C1s, C2, C3, C4, C5, C6, C7, C8, and C9, in serum, other body fluids, and tissues. Complement is a group of serum proteins which destroy infectious agents. Measurements of these proteins aids in the diagnosis of immunologic disorders, especially those associated with deficiencies of complement components.
(b) Classification. Class II (performance standards).
[47 FR 50823, Nov. 9, 1982, as amended at 53 FR 11253, Apr. 6, 1988]
|
|
|
Sec. 866.5250 Complement C1 inhibitor (inactivator) immunological test system.
|
|
(a) Identification. A complement C1 inhibitor (inactivator) immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the complement C1 inhibitor (a plasma protein) in serum. Complement C1 inhibitor occurs normally in plasma and blocks the action of the C1 component of complement (a group of serum proteins which destroy infectious agents). Measurement of complement C1 inhibitor aids in the diagnosis of hereditary angioneurotic edema (increased blood vessel permeability causing swelling of tissues) and a rare form of angioedema associated with lymphoma (lymph node cancer).
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5260 Complement C3b inactivator immunological test system.
|
|
(a) Identification. A complement C3b inactivator immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the complement C3b inactivator (a plasma protein) in serum. Complement is a group of serum proteins that destroy infectious agents. Measurement of complement C3b inactivator aids in the diagnosis of inherited antibody dysfunction.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5270 C-reactive protein immunological test system.
|
|
(a) Identification. A C-reactive protein immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the C-reactive protein in serum and other body fluids. Measurement of C-reactive protein aids in evaluation of the amount of injury to body tissues.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5320 Properdin factor B immunological test system.
|
|
(a) Identification. A properdin factor B immunological test system is a device that consists of the reagents used to measure by immunochemical techniques properdin factor B in serum and other body fluids. The deposition of properdin factor B in body tissues or a corresponding depression in the amount of properdin factor B in serum and other body fluids is evidence of the involvement of the alternative to the classical pathway of activation of complement (a group of plasma proteins which cause the destruction of cells which are foreign to the body). Measurement of properdin factor B aids in the diagnosis of several kidney diseases, e.g., chronic glomerulonephritis (inflammation of the glomeruli of the kidney), lupus nephritis (kidney disease associated with a multisystem autoimmune disease, systemic lupus erythematosus), as well as several skin diseases, e.g., dermititis herpetiformis (presence of vesicles on the skin that burn and itch), and pemphigus vulgaris (large vesicles on the skin). Other diseases in which the alternate pathway of complement activation has been implicated include rheumatoid arthritis, sickle cell anemia, and gram-negative bacteremia.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 63 FR 59227, Nov. 3, 1998]
|
|
|
Sec. 866.5330 Factor XIII, A, S, immunological test system.
|
|
(a) Identification. A factor XIII, A, S, immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the factor XIII (a bloodclotting factor), in platelets (A) or serum (S). Measurements of factor XIII, A, S, aid in the diagnosis and treatment of certain bleeding disorders resulting from a deficiency of this factor.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9. This exemption does not apply to factor deficiency tests classified under § 864.7290 of this chapter.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5340 Ferritin immunological test system.
|
|
(a) Identification. A ferritin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the ferritin (an iron-storing protein) in serum and other body fluids. Measurements of ferritin aid in the diagnosis of diseases affecting iron metabolism, such as hemochromatosis (iron overload) and iron deficiency amemia.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5350 Fibrinopeptide A immunological test system.
|
|
(a) Identification. A fibrinopeptide A immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the fibrinopeptide A (a blood-clotting factor) in plasma and other body fluids. Measurement of fibrinopeptide A may aid in the diagnosis and treatment of certain blood-clotting disorders.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5360 Cohn fraction IV immunological test system.
|
|
(a) Identification. A Cohn fraction IV immunological test system is a device that consists of or measures that fraction of plasma proteins, predominantly alpha- and beta- globulins, used as a raw material for the production of pure alpha- or beta- globulins. Measurement of specific alpha- or beta- globulins aids in the diagnosis of many diseases, such as Wilson's disease (an inherited disease affecting the liver and brain), Tangier's disease (absence of alpha- 1-lipoprotein), malnutrition, iron deficiency anemia, red blood cell disorders, and kidney disease.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982; 47 FR 56846, Dec. 21, 1982, as amended at 59 FR 63007, Dec. 7, 1994; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5370 Cohn fraction V immunological test system.
|
|
(a) Identification. A Cohn fraction V immunological test system is a device that consists of or measures that fraction of plasma containing predominantly albumin (a plasma protein). This test aids in the diagnosis of diseases where albumin levels may be depressed, e.g., nephrosis (disease of the kidney), proteinuria (protein in the urine), gastroenteropathy (disease of the stomach and small intestine), rheumatoid arthritis, and viral hepatitis.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 59 FR 63007, Dec. 7, 1994; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5380 Free secretory component immunological test system.
|
|
(a) Identification. A free secretory component immunological test system is a device that consists of the reagents used to measure by immunochemical techniques free secretory component (normally a portion of the secretory IgA antibody molecule) in body fluids. Measurement of free secretory component (protein molecules) aids in the diagnosis or repetitive lung infections and other hypogammaglobulinemic conditions (low antibody levels).
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 63 FR 59227, Nov. 3, 1998]
|
|
|
Sec. 866.5400 Alpha-globulin immunological test system.
|
|
(a) Identification. An alpha- globulin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the alpha- globulin (a serum protein) in serum and other body fluids. Measurement of alpha- globulin may aid in the diagnosis of inflammatory lesions, infections, severe burns, and a variety of other conditions.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5420 Alpha-1-glycoproteins immunological test system.
|
|
(a) Identification. An alpha- 1-glycoproteins immunological test system is a device that consists of the reagents used to measure by immunochemical techniques alpha- 1-glycoproteins (a group of plasma proteins found in the alpha- 1 group when subjected to electrophoresis) in serum and other body fluids. Measurement of specific alpha- 1-glycoproteins may aid in the diagnosis of collagen (connective tissue) disorders, tuberculosis, infections, extensive malignancy, and diabetes.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5425 Alpha-2-glycoproteins immunological test system.
|
|
(a) Identification. An alpha -2-glycoproteins immunolgical test system is a device that consists of the reagents used to measure by immunochemical techniques the alpha -2-glycoproteins (a group of plasma proteins found in the alpha- 2 group when subjected to electrophoresis) in serum and other body fluids. Measurement of alpha -2-glycoproteins aids in the diagnosis of some cancers and genetically inherited deficiencies of these plasma proteins.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5430 Beta-2-glycoprotein I immunological test system.
|
|
(a) Identification. A beta -2-glycoprotein I immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the beta -2-glycoprotein I (a serum protein) in serum and other body fluids. Measurement of beta -2-glycoprotein I aids in the diagnosis of an inherited deficiency of this serum protein.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5440 Beta-2-glycoprotein III immunological test system.
|
|
(a) Identification. A beta -2-glycoprotein III immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the beta -2-glycoprotein III (a serum protein) in serum and other body fluids. Measurement of beta -2-glycoprotein III aids in the diagnosis of an inherited deficiency of this serum protein and a variety of other conditions.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5460 Haptoglobin immunological test system.
|
|
(a) Identification. A haptoglobin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the haptoglobin (a protein that binds hemoglobin, the oxygen-carrying pigment in red blood cells) in serum. Measurement of haptoglobin may aid in the diagnosis of hemolytic diseases (diseases in which the red blood cells rupture and release hemoglobin) related to the formation of hemoglobin-haptoglobin complexes and certain kidney diseases.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 63 FR 59227, Nov. 3, 1998]
|
|
|
Sec. 866.5470 Hemoglobin immunological test system.
|
|
(a) Indentification. A hemoglobin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the different types of free hemoglobin (the oxygen-carrying pigment in red blood cells) in blood, urine, plasma, or other body fluids. Measurements of free hemoglobin aid in the diagnosis of various hematologic disorders, such as sickle cell anemia, Fanconi's anemia (a rare inherited disease), aplastic anemia (bone marrow does not produce enough blood cells), and leukemia (cancer of the blood-forming organs).
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 84 FR 71800, Dec. 30, 2019]
|
|
|
Sec. 866.5490 Hemopexin immunological test system.
|
|
(a) Indentification. A hemopexin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the hemopexin (a serum protein that binds heme, a component of hemoglobin) in serum. Measurement of hemopexin aids in the diagnosis of various hematologic disorders, such as hemolytic anemia (anemia due to shortened in vivo survival of mature red blood cells and inability of the bone marrow to compensate for their decreased life span) and sickle cell anemia.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 63 FR 59227, Nov. 3, 1998]
|
|
|
Sec. 866.5500 Hypersensitivity pneumonitis immunological test system.
|
|
(a) Identification. A hypersensitivity pneumonitis immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the immunoglobulin antibodies in serum which react specifically with organic dust derived from fungal or animal protein sources. When these antibodies react with such dusts in the lung, immune complexes precipitate and trigger an inflammatory reaction (hypersensitivity pneumonitis). Measurement of these immunoglobulin G antibodies aids in the diagnosis of hypersensitivity pneumonitis and other allergic respiratory disorders.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5510 Immunoglobulins A, G, M, D, and E immunological test system.
|
|
(a) Identification. An immunoglobulins A, G, M, D, and E immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the immunoglobulins A, G, M, D, an E (serum antibodies) in serum. Measurement of these immunoglobulins aids in the diagnosis of abnormal protein metabolism and the body's lack of ability to resist infectious agents.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5520 Immunoglobulin G (Fab fragment specific) immunological test system.
|
|
(a) Identification. An immunoglobulin G (Fab fragment specific) immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the Fab antigen-binding fragment resulting from breakdown of immunoglobulin G antibodies in urine, serum, and other body fluids. Measurement of Fab fragments of immunoglobulin G aids in the diagnosis of lymphoproliferative disorders, such as multiple myeloma (tumor of bone marrow cells), Waldenstrom's macroglobulinemia (increased immunoglobulin production by the spleen and bone marrow cells), and lymphoma (tumor of the lymphoid tissues).
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 61 FR 1119, Jan. 16, 1996; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5530 Immunoglobulin G (Fc fragment specific) immunological test system.
|
|
(a) Identification. An immunoglobulin G (Fc fragment specific) immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the Fc (carbohydrate containing) fragment of immunoglobulin G (resulting from breakdown of immunoglobulin G antibodies) in urine, serum, and other body fluids. Measurement of immunoglobulin G Fc fragments aids in the diagnosis of plasma cell antibody-forming abnormalities, e.g., gamma heavy chain disease.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 61 FR 1119, Jan. 16, 1996; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5540 Immunoglobulin G (Fd fragment specific) immunological test system.
|
|
(a) Identification. An immunoglobulin G (Fd fragment specific) immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the amino terminal (antigen-binding) end (Fd fragment) of the heavy chain (a subunit) of the immunoglobulin antibody molecule in serum. Measurement of immunoglobulin G Fd fragments aids in the diagnosis of plasma antibody-forming cell abnormalities.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 59 FR 63007, Dec. 7, 1994; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5550 Immunoglobulin (light chain specific) immunological test system.
|
|
(a) Identification. An immunoglobulin (light chain specific) immunological test system is a device that consists of the reagents used to measure by immunochemical techniques both kappa and lambda types of light chain portions of immunoglobulin molecules in serum, other body fluids, and tissues. In some disease states, an excess of light chains are produced by the antibody-forming cells. These free light chains, unassociated with gamma globulin molecules, can be found in a patient's body fluids and tissues. Measurement of the various amounts of the different types of light chains aids in the diagnosis of multiple myeloma (cancer of antibody-forming cells), lymphocytic neoplasms (cancer of lymphoid tissue), Waldenstrom's macroglobulinemia (increased production of large immunoglobulins), and connective tissue diseases such as rheumatoid arthritis or systemic lupus erythematosus.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5560 Lactic dehydrogenase immunological test system.
|
|
(a) Identification. A lactic dehydrogenase immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the activity of the lactic dehydrogenase enzyme in serum. Increased levels of lactic dehydrogenase are found in a variety of conditions, including megaloblastic anemia (decrease in the number of mature red blood cells), myocardial infarction (heart disease), and some forms of leukemia (cancer of the blood-forming organs). However, the diagnostic usefulness of this device is limited because of the many conditions known to cause increased lactic dehydrogenase levels.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5570 Lactoferrin immunological test system.
|
|
(a) Identification. A lactoferrin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the lactoferrin (an iron-binding protein with the ability to inhibit the growth of bacteria) in serum, breast milk, other body fluids, and tissues. Measurement of lactoferrin may aid in the diagnosis of an inherited deficiency of this protein.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2312, Jan. 14, 2000]
|
|
|
Sec. 866.5580 Alpha-1-lipoprotein immunological test system.
|
|
(a) Identification. An alpha -1-lipoprotein immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the alpha- 1-lipoprotein (high-density lipoprotein) in serum and plasma. Measurement of alpha- 1-lipoprotein may aid in the diagnosis of Tangier disease (a hereditary disorder of fat metabolism).
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5590 Lipoprotein X immunological test system.
|
|
(a) Identification. A lipoprotein X immunological test system is a device that consists of the reagents used to measure by immunochemical techniques lipoprotein X (a high-density lipoprotein) in serum and other body fluids. Measurement of lipoprotein X aids in the diagnosis of obstructive liver disease.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2313, Jan. 14, 2000]
|
|
|
Sec. 866.5600 Low-density lipoprotein immunological test system.
|
|
(a) Identification. A low-density lipoprotein immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the low-density lipoprotein in serum and other body fluids. Measurement of low-density lipoprotein in serum may aid in the diagnosis of disorders of lipid (fat) metabolism and help to identify young persons at risk from cardiovascular diseases.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5620 Alpha-2-macroglobulin immunological test system.
|
|
(a) Identification. An alpha -2-macroglobulin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the alpha -2-macroglobulin (a serum protein) in plasma. Measurement of alpha -2-macroglobulin may aid in the diagnosis of blood-clotting or clot lysis disorders.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 84 FR 71800, Dec. 30, 2019]
|
|
|
Sec. 866.5630 Beta-2-microglobulin immunological test system.
|
|
(a) Identification. A beta -2-microglobulin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques beta -2-microglobulin (a protein molecule) in serum, urine, and other body fluids. Measurement of beta -2-microglobulin aids in the diagnosis of active rheumatoid arthritis and kidney disease.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 84 FR 71800, Dec. 30, 2019]
|
|
|
Sec. 866.5640 Infectious mononucleosis immunological test system.
|
|
(a) Identification. An infectious mononucleosis immunological test system is a device that consists of the reagents used to measure by immunochemical techniques heterophile antibodies frequently associated with infectious mononucleosis in serum, plasma, and other body fluids. Measurements of these antibodies aid in the diagnosis of infectious mononucleosis.
(b) Classification. Class II (performance standards).
[47 FR 50823, Nov. 9, 1982; 47 FR 56846, Dec. 21, 1982]
|
|
|
Sec. 866.5660 Multiple autoantibodies immunological test system.
|
|
(a) Identification. A multiple autoantibodies immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the autoantibodies (antibodies produced against the body's own tissues) in serum and other body fluids. Measurement of multiple autoantibodies aids in the diagnosis of autoimmune disorders (disease produced when the body's own tissues are injured by autoantibodies).
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5665 Aquaporin-4 autoantibody immunological test system.
|
|
(a) Identification. An Aquaporin-4 autoantibody immunological test system is a device that consists of reagents used to measure by immunochemical techniques autoantibodies in human serum samples that react with Aquaporin-4 (AQP4Ab). The measurements aid in the diagnosis of neuromyelitis optica (NMO) and neuromyelitis optica spectrum disorders (NMOSD) in conjunction with other clinical, laboratory, and radiological (e.g., magnetic resonance imaging) findings.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) Premarket notification submissions must include the following information:
(i) A detailed device description including:
(A) A detailed description of all components including all required ancillary reagents in the test;
(B) If applicable, a detailed description of instrumentation and equipment, including illustrations or photographs of non-standard equipment or manuals;
(C) If applicable, detailed documentation of the device software, including, but not limited to, standalone software applications and hardware-based devices that incorporate software;
(D) A detailed description of appropriate internal and external quality controls that are recommended or provided. The description must identify those control elements that are incorporated into the specified testing procedures;
(E) Detailed specifications for sample collection, processing, and storage;
(F) A detailed description of methodology and assay procedure;
(G) A description of how the assay cutoff (the medical decision point between positive and negative) was established and validated as well as supporting data; and
(H) Detailed specification of the criteria for test results interpretation and reporting.
(ii) Detailed information demonstrating the performance characteristics of the device, including:
(A) Device precision/reproducibility data generated from within-run, between-run, between-day, between-lot, between-site, and total precision for multiple nonconsecutive days, as applicable. A well characterized panel of patient samples or pools from the indicated population that covers the device measuring range must be used.
(B) Device linearity data generated from samples covering the device measuring range, if applicable.
(C) Information on traceability to a reference material and description of value assignment of calibrators and controls, if applicable.
(D) Device analytical sensitivity data, including limit of blank, limit of detection, and limit of quantitation, if applicable.
(E) Device analytical specificity data, including interference by endogenous and exogenous substances, as well as cross-reactivity with samples derived from patients with other autoimmune diseases or conditions.
(F) Device instrument carryover data, when applicable.
(G) Device stability data, including real-time stability under various storage times and temperatures.
(H) Specimen stability data, including stability under various storage times, temperatures, freeze-thaw, and transport conditions, where appropriate.
(I) Method comparison data generated by comparison of the results obtained with the device to those obtained with a legally marketed predicate device with similar indications of use. A well-characterized panel of patient samples from the indicated population covering the device measuring range must be used.
(J) Specimen matrix comparison data, if more than one specimen type or anticoagulant can be tested with the device. Samples used for comparison must be from well-characterized patient samples covering the device measuring range.
(K) Clinical performance must be established by comparing data generated by testing samples from the indicated population and the differential diagnosis or non-target disease groups with the device to the clinical diagnostic standard.
(1 ) The diagnosis of NMO and NMOSD must be based on clinical findings, laboratory tests (e.g., serological tests), and radiological tests (e.g., magnetic resonance imaging).
(2 ) The differential diagnosis or non-target disease group must include the applicable diseases or conditions, including but not be limited to the following: Multiple sclerosis, stroke, Lyme disease, shingles, syphilis, human immunodeficiency virus, hepatitis B, tuberculosis, Srgen's syndrome, systemic lupus erythematous, systemic vasculitis, sarcoidosis, Graves' disease, Hashimoto's disease, Type I diabetes, rheumatoid arthritis, Addison's disease, and myasthenia gravis.
(3 ) Diagnosis of diseases or conditions for the differential or non-target disease groups must be based on established diagnostic criteria and clinical evaluation.
(4 ) For all samples, the diagnostic clinical criteria and the demographic information must be collected and provided.
(5 ) The clinical validation results must demonstrate clinical sensitivity and clinical specificity for the test values based on the presence or absence of NMO and NMOSD.
(6 ) The data must be summarized in tabular format comparing the interpretation of results to the disease status.
(L) Expected/reference values generated by testing an adequate number of samples from apparently healthy normal individuals.
(iii) Identification of risk mitigation elements used by the device, including description of all additional procedures, methods, and practices incorporated into the directions for use that mitigate risks associated with testing.
(2) The device's 21 CFR 809.10(b) compliant labeling must include warnings relevant to the device including:
(i) A warning statement that reads "The device is for use by laboratory professionals in a clinical laboratory setting"; and
(ii) A warning statement that reads "The device is not to be used as a stand-alone device but as an adjunct to other clinical information. A diagnosis of Neuromyelitis Optica (NMO) and Neuromyelitis Optica Spectrum Disorders (NMOSD) should not be made on a single test result. The clinical symptoms, results from physical examination, laboratory tests (e.g., serological tests), and radiological tests (e.g. Magnetic Resonance Imaging), when appropriate, should always be taken into account when considering the diagnosis of NMO and NMOSD."
(3) The device's 21 CFR 809.10(b) compliant labeling must include a detailed description of the protocol and performance studies performed in accordance with paragraph (b)(1)(ii) of this section and a summary of the results.
[82 FR 50076, Oct. 30, 2017]
|
|
|
Sec. 866.5670 Zinc transporter 8 autoantibody immunological test system.
|
|
(a) Identification. A zinc transporter 8 autoantibody immunological test system is a device that consists of reagents used to measure, by immunochemical techniques, the autoantibodies in human serum samples that react with Zinc Transporter 8 (ZnT8). The measurements aid in the diagnosis of Type 1 diabetes mellitus (autoimmune mediated diabetes) in conjunction with other clinical and laboratory findings.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) Premarket notification submissions must include the following information:
(i) A detailed description of the device that includes:
(A) A detailed description of all components in the test system, including a description of the assay components in the kit and all required ancillary reagents;
(B) A detailed description of instrumentation and equipment, and illustrations or photographs of non-standard equipment or methods if applicable;
(C) Detailed documentation of the device software, including, but not limited to, standalone software applications and hardware-based devices that incorporate software where applicable;
(D) A detailed description of appropriate internal and external quality controls that are recommended or provided. The description must identify those control elements that are incorporated into the recommended testing procedures;
(E) Detailed specifications for sample collection, processing, and storage;
(F) A detailed description of methodology and assay procedure; and
(G) Detailed specification of the criteria for test results interpretation and reporting.
(ii) Information that demonstrates the performance characteristics of the device, including:
(A) Device precision/reproducibility data generated from within-run, between-run, between-day, between-lot, between-operator, between-instruments, between-site, and total precision for multiple nonconsecutive days as applicable. A well characterized panel of patient samples or pools from the intended use population that covers the device measuring range must be used;
(B) Device linearity data generated from patient samples covering the assay measuring range if applicable;
(C) Information on traceability to a reference material and description of value assignment of calibrators and controls if applicable;
(D) Device analytical sensitivity data, including limit of blank, limit of detection and limit of quantitation if applicable;
(E) Device analytical specificity data, including interference by endogenous and exogenous substances, as well as cross-reactivity with samples derived from patients with other autoimmune diseases or conditions;
(F) Device instrument carryover data when applicable;
(G) Device stability data including real-time stability under various storage times and temperatures;
(H) Specimen stability data, including stability under various storage times, temperatures, freeze-thaw, and transport conditions where appropriate;
(I) Method comparison data generated by comparison of the results obtained with the device to those obtained with a legally marketed predicate device with similar indication of use. Patient samples from the intended use population covering the device measuring range must be used;
(J) Specimen matrix comparison data if more than one specimen type or anticoagulant can be tested with the device. Samples used for comparison must be from patient samples covering the device measuring range;
(K) A description of how the assay cut-off (the medical decision point between positive and negative) was established and validated as well as supporting data;
(L) Clinical performance must be established by comparing data generated by testing samples from the intended use population and the differential diagnosis groups with the device to the clinical diagnostic standard. The diagnosis of Type 1 diabetes mellitus must be based on clinical history, physical examination, and laboratory tests, such as one or more pancreatic or insulin autoantibody test. Because the intended use population for Type 1 diabetes mellitus includes subjects less than 18 years old, samples from representative numbers of these subjects must be included. Representative numbers of samples from all age strata must also be included. The differential diagnosis groups must include, but not be limited to the following: Type 2 diabetes mellitus; metabolic syndrome; latent autoimmune diabetes in adults; other autoimmune diseases such as celiac disease (without a concomitant diagnosis of Type 1 diabetes mellitus), systemic lupus erythematosus, rheumatoid arthritis, and Hashimoto's thyroiditis; infection; renal disease; and testicular cancer. Diseases for the differential groups must be based on established diagnostic criteria and clinical evaluation. For all samples, the diagnostic clinical criteria and the demographic information must be collected and provided. The clinical validation results must demonstrate clinical sensitivity and clinical specificity for the test values based on the presence or absence of Type 1 diabetes mellitus. The data must be summarized in tabular format comparing the interpretation of results to the disease status; and
(M) Expected/reference values generated by testing an adequate number of samples from apparently healthy normal individuals.
(iii) Identification of risk mitigation elements used by the device, including description of all additional procedures, methods, and practices incorporated into the directions for use that mitigate risks associated with testing.
(2) Your 21 CFR 809.10(a) compliant label and 21 CFR 809.10(b) compliant labeling must include warnings relevant to the assay including:
(i) A warning statement that reads, "The device is for use by laboratory professionals in a clinical laboratory setting";
(ii) A warning statement that reads, "The test is not a stand-alone test but an adjunct to other clinical information. A diagnosis of Type 1 diabetes mellitus should not be made on a single test result. The clinical symptoms, results on physical examination, and laboratory tests (e.g., serological tests), when appropriate, should always be taken into account when considering the diagnosis of Type 1 diabetes mellitus and Type 2 diabetes mellitus";
(iii) A warning statement that reads, "Absence of Zinc T8 autoantibody does not rule out a diagnosis of Type 1 diabetes mellitus"; and
(iv) A warning statement that reads, "The assay has not been demonstrated to be effective for monitoring the stage of disease or its response to treatment."
(3) Your 21 CFR 809.10(b) compliant labeling must include a description of the protocol and performance studies performed in accordance with paragraph (b)(1)(ii) of this section and a summary of the results.
[82 FR 49103, Oct. 20, 2017]
|
|
|
Sec. 866.5680 Myoglobin immunological test system.
|
|
(a) Identification. A myoglobin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the myoglobin (an oxygen storage protein found in muscle) in serum and other body fluids. Measurement of myoglobin aids in the rapid diagnosis of heart or renal disease.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5700 Whole human plasma or serum immunological test system.
|
|
(a) Identification. A whole human plasma or serum immunological test system is a device that consists of reagents used to measure by immunochemical techniques the proteins in plasma or serum. Measurements of proteins in plasma or serum aid in the diagnosis of any disease concerned with abnormal levels of plasma or serum proteins, e.g., agammaglobulinemia, allergies, multiple myeloma, rheumatoid vasculitis, or hereditary angioneurotic edema.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 59 FR 63007, Dec. 7, 1994; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5715 Plasminogen immunological test system.
|
|
(a) Identification. A plasminogen immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the plasminogen (an inactive substance from which plasmin, a blood-clotting factor, is formed) in serum, other body fluids, and tissues. Measurement of plasminogen levels may aid in the diagnosis of fibrinolytic (blood-clotting) disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2313, Jan. 14, 2000]
|
|
|
Sec. 866.5735 Prothrombin immunological test system.
|
|
(a) Identification. A prothrombin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the prothrombin (clotting factor II) in serum. Measurements of the amount of antigenically competent (ability to react with protein antibodies) prothrombin aid in the diagnosis of blood-clotting disorders.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9. This exemption does not apply to multipurpose systems for in vitro coagulation studies classified under § 864.5425 of this chapter or prothrombin time tests classified under § 864.7750 of this chapter.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2313, Jan. 14, 2000]
|
|
|
Sec. 866.5750 Radioallergosorbent (RAST) immunological test system.
|
|
(a) Identification. A radioallergosorbent immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the allergen antibodies (antibodies which cause an allergic reaction) specific for a given allergen. Measurement of specific allergen antibodies may aid in the diagnosis of asthma, allergies, and other pulmonary disorders.
(b) Classification. Class II (special controls). The device, when intended to detect any of the allergens included in Table 1 in this paragraph, is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
Table 1 - Class II Exempt Allergens Under § 866.5750 - Radioallergosorbent (RAST) Immunological Test Systems
| Allergen code
| Allergen product
| Source
(taxonomical name)
|
|---|
| Grass Pollens
| | g1 | Sweet vernal grass | Anthoxanthum odoratum.
| | g3 | Cocksfoot grass, Orchard grass | Dactylis glomerata.
| | g4 | Meadow fescue | Festuca elatior.
| | g5 | Rye-grass (perennial rye grass) | Lolium perenne.
| | g7 | Common reed (common reed grass) | Phragmites communis.
| | g8 | Meadow grass, Kentucky blue (June grass) | Poa pratensis.
| | g9 | Redtop, Bentgrass | Agrostis stolonifera, Agrostis gigantea (Agrostis alba).
| | g11 | Brome grass | Bromus inermis.
| | g12 | Cultivated rye (cultivated rye grass) | Secale cereale.
| | g13 | Velvet grass | Holcus lanatus.
| | g14 | Cultivated oat (cultivated oat grass) | Avena sativa.
| | g15 | Cultivated wheat (cultivated wheat grass) | Triticum aestivum (Triticum spp.).
| | g16 | Meadow foxtail (meadow foxtail grass) | Alopecurus pratensis.
| | g17 | Bahia grass | Paspalum notatum.
| | g24 | Wheat grass, Western | Agropyron smithii (Elymus smithii).
| | g30 | Bluegrass, annual | Poa annua.
| | g70 | Wild rye grass | Elymus triticoides Elymus condensatus.
| | g71 | Canary grass | Phalaris arundinacea.
| | g201 | Barley, cultivated | Hordeum vulgare.
| | g202 | Maize, corn (cultivated corn) | Zea mays.
| | g203 | Salt grass | Distichlis spicata.
| | g204 | False oat-grass | Arrhenatherum elatius.
| | g216 | Cyn d 1 | Cynodon dactylon.
| | g701 | Phl p 1.0102, Phl p 5.0101 | Phleum pratense.
| | g702 | Phl p 7.0101 | Phleum pratense.
| | g703 | Phl p 12.0101 | Phleum pratense.
| | Weed Pollens
| | w2 | Western ragweed | Ambrosia psilostachya.
| | w4 | False ragweed | Ambrosia acanthicarpa (Franseria acanthicarpa).
| | w5 | Wormwood | Artemisia absinthium Artemisia annua.
| | w6 | Mugwort | Artemisia vulgaris.
| | w7 | Marguerite, ox-eye daisy | Chrysanthemum leucanthemum.
| | w8 | Dandelion | Taraxacum vulgare, Taraxacum officinale.
| | w9 | Plantain (English), Ribwort | Plantago lanceolata.
| | w10 | Goosefoot, lamb's quarters | Chenopodium album.
| | w11 | Saltwort (prickly), Russian thistle | Salsola kali (Salsola pestifer).
| | w12 | Goldenrod | Solidago virgaurea (Solidago spp.).
| | w13 | Cocklebur, common | Xanthium commune.
| | w14 | Common pigweed (rough pigweed) | Amaranthus retroflexus.
| | w15 | Scale, Lenscale | Atriplex lentiformis.
| | w16 | Rough marsh elder | Iva ciliate, Iva annua.
| | w17 | Firebush (Kochia) | Kochia scoparia.
| | w18 | Sheep sorrel | Rumex acetosella.
| | w19 | Wall pellitory | Parietaria officinalis.
| | w20 | Nettle (Common stinging nettle) | Urtica dioica.
| | w21 | Wall pellitory | Parietaria judaica.
| | w22 | Japanese hop (careless weed) | Humulus japonicas (Humulus scandens).
| | w23 | Yellow dock, Yellow dockweed | Rumex crispus.
| | w24 | Spiny pigweed | Amaranthus spinosus.
| | w27 | Carnation | Dianthus spp.
| | w28 | Rose | Rosa rugosa.
| | w33 | Clover | Trifolium pratense.
| | w35 | Mexican tea | Chenopodium ambrosioides.
| | w36 | Rabbit bush | Ambrosia deltoidea (Franseria deltoides).
| | w37 | Salt bush, annual | Atriplex wrightii.
| | w39 | Water hemp, Western | Amaranthus rudis (Acnida tamariscina).
| | w41 | Burrobrush | Hymenoclea salsola.
| | w42 | Poverty weed | Baccharis neglecta.
| | w43 | Common sagebrush | Artemisia tridentata.
| | w45 | Alfalfa | Medicago sativa.
| | w46 | Dog fennel | Eupatorium capillifolium.
| | w53 | Geranium | Geranium spp.
| | w67 | Groundsel bush | Baccharis halimifolia.
| | w69 | Iodine bush | Allenrolfea occidentalis.
| | w70 | Ragweed, slender | Ambrosia confertiflora.
| | w75 | Wing scale (wingscale) | Atriplex canescens.
| | w82 | Careless weed | Amaranthus palmeri, Amaranthus hybridus.
| | w90 | Japanese hop | Humulus japonicas (Humulus scandens).
| | w203 | Rape (rape pollen) | Brassica napus.
| | w204 | Sunflower | Helianthus annuus.
| | w206 | Camomile | Matricaria chamomilla.
| | w207 | Lupin | Lupinus spp.
| | w210 | Sugar-beet | Beta vulgaris.
| | w211 | Par j 2.0101 | Parietaria judaica.
| | w231 | Art v 1 | Artemisia vulgaris (Mugwort).
| | w232 | Sal k 1 | Salsola kali.
| | w233 | Art v 3 | Artemisa vulgaris (LTP, Mugwort).
| | w234 | Pla l 1 | Plantago lanceolata.
| | w235 | Che a 1.0101 | Chenopodium album.
| | w236 | Mer a 1.0101 | Mercurialis annua.
| | a753 | Art v 1 | Artemisia vulgaris (Mugwort weed).
| | Tree Pollens
| | t1 | Box-elder (Maple) | Acer negundo, Acer saccharum.
| | t2 | Gray alder, speckled alder (alder) | Alnus incana.
| | t4 | Hazel, hazelnut | Corylus avellana, Corylus americana.
| | t5 | American beech (beech) | Fagus grandifolia (Fagus americana).
| | t6 | Mountain juniper, Mountain cedar | Juniperus ashei (Juniperus sabinoides).
| | t8 | Elm | Ulmus americana.
| | t9 | Olive | Olea europaea.
| | t10 | Walnut | Juglans californica, Juglans nigra.
| | t11 | Maple leaf sycamore, London plane, Plane tree | Platanus acerifolia.
| | t61 | Sycamore | Platanus occidentalis.
| | t12 | Willow | Salix caprea, Salix nigra.
| | t14 | Cottonwood (Eastern Cottonwood/Black Cottonwood) | Populus deltoides.
| | t15 | White ash | Fraxinus americana.
| | t16 | White pine | Pinus strobus.
| | t18 | Eucalyptus, gum-tree | Eucalyptus globulus (Eucalyptus spp.).
| | t19/t26 | Acacia | Acacia longifolia (Acacia spp.).
| | t20 | Mesquite | Prosopis glandulosa/Prosopis juliflora.
| | t21 | Melaleuca, cajeput tree | Melaleuca quinquenervia (Melaleuca leucadendron).
| | t22 | Pecan, hickory | Carya illinoinensis (Carya pecan).
| | t23 | Italian/Mediterranean/funeral cypress | Cupressus sempervirens.
| | t24 | Japanese cypress | Chamaecyparis obtusa (Chamaecyparis spp.).
| | t25 | Ash | Fraxinus excelsior.
| | t27 | Maple, red | Acer rubrum.
| | t29 | Acacia | Acacia spp.
| | t30 | Birch, white | Betula populifolia.
| | t32 | Willow, black | Salix nigra.
| | t33 | Ash, Arizona | Fraxinus velutina.
| | t35 | Cedar, salt | Tamarix gallica.
| | t37 | Bald cypress (white bald cypress) | Taxodium distichum.
| | t38 | Elm, Chinese/Siberian | Ulmus pumila.
| | t40 | Hazelnut tree | Corylus americana.
| | t41 | White hickory | Carya alba (Carya tomentosa).
| | t42 | Oak, red | Quercus rubra.
| | t43 | Loblolly pine | Pinus taeda.
| | t44 | Hackberry | Celtis occidentalis.
| | t45 | Cedar elm | Ulmus crassifolia.
| | t47 | Juniper, one seed | Juniperus monosperma.
| | t48 | Pine, lodgepole | Pinus contorta.
| | t49 | Pine, ponderosa | Pinus ponderosa.
| | t50 | Beech, European | Fagus sylvatica.
| | t51 | Tree of Heaven | Ailanthus altissima.
| | t52 | Western white pine | Pinus monticola.
| | t54 | Russian olive | Elaeagnus angustifolia.
| | t55 | Scotch broom | Cytisus scoparius.
| | t56 | Bayberry | Myrica cerifera.
| | t57 | Red cedar | Juniperus virginiana.
| | t60 | Western juniper | Juniperus occidentalis.
| | t61 | Sycamore | Platanus occidentalis.
| | t70 | Mulberry (white mulberry) | Morus alba.
| | t71 | Red mulberry | Morus rubra.
| | t72 | Queen palm | Arecastrum romanzoffiamon.
| | t73 | Australian pine | Casuarina equisetifolia.
| | t77 | Oak mix (red, white, black) | Quercus spp.
| | t80 | Japanese cypress | Chamaecyparis obtusa.
| | t81 | Japanese alder | Alnus japonica.
| | t83 | Mango tree | Mangifera indica.
| | t90 | Walnut, black | Juglans nigra.
| | t96 | Poplar, white (poplar) | Populus alba.
| | t103/t218 | Virginia live oak (live oak) | Quercus virginiana.
| | t105 | Pepper tree | Schinus molle.
| | t110 | Orange tree | Citrus sinensis.
| | t201 | Spruce, Norway spruce | Picea abies (Picea excelsa).
| | t202 | Alder, smooth | Alnus incana spp. Rugosa (Alnus rugosa).
| | t203 | Horse chestnut | Aesculus hippocastanum.
| | t205 | Elder | Sambucus nigra.
| | t206 | Chestnut | Castanea sativa.
| | t207 | Douglas fir | Pseudotsuga menziesii (Pseudotsuga taxifolia).
| | t208 | Linden | Tilia cordata.
| | t209 | Horn beam | Carpinus betulus.
| | t210 | Privet | Ligustrum vulgare.
| | t211 | Sweet gum | Liquidambar styraciflua.
| | t212 | Cedar | Libocedrus decurrens.
| | t213 | Pine | Pinus radiata.
| | t214 | Date palm | Phoenix canariensis.
| | t215 | Lilac | Syringa vulgaris.
| | t217 | Pepper tree | Schinus molle.
| | t217 | Red alder | Alnus rubra.
| | t218 | Virginia live oak | Quercus virginiana.
| | t218 | Bayberry (bayberry/sweet gale) | Myrica gale.
| | t219 | Palo verde | Cercidium floridum.
| | t219 | Red cedar | Juniperus virginiana.
| | t220 | Bet v 4 | Betula verrucosa (Birch).
| | t221 | Bet v 2.0101, Bet v 4 | Betula verrucosa (Birch).
| | t222 | Cypress (Arizona cypress) | Cupressus arizonica.
| | t223 | Oil palm | Elaeis guineensis.
| | t224 | Ole e 1 | Olea europaea.
| | t225 | Bet v 6 | Betula verrucosa (Birch).
| | t226 | Cup a 1 | Cupressus arizonica.
| | t227 | Ole e 7 | Olea Europaea.
| | t228 | Aspen, quaking | Populus tremuloides.
| | t229 | Eastern hemlock | Tsuga canadensis.
| | t230 | Redwood (sequoia) | Sequoia sempervirens.
| | t232 | Pussy willow | Salix discolor.
| | t240 | Ole e 9.0101 | Olea Europaea.
| | t241 | Pla a 1.0101 | Platanus acerifolia.
| | t242 | Pla a 2 | Platanus acerifolia.
| | t243 | Pla a 3.0101 | Platanus acerifolia.
| | t244 | Cor a 1.0103 | Corylus avellana.
| | t245 | Aln g 1.0101 | Alnus glutinosa.
| | t246 | Cry j 1 | Cryptomeria japonica.
| | t280 | Locust tree | Robinia pseudoacacia.
| | t401 | Brazilian peppertree | Schinus terebinthifolius.
| | t402 | Mastic tree | Pistacia lentiscus.
| | t404 | Tree of heaven | Ailanthus altissima.
| | t406 | Date palm | Phoenix dactylifera.
| | a482 | Ole e 1 | Olea europaea (Olive Oil).
| | Mites
| | d207 | Blo t 5.0101 | Blomia tropicalis.
| | d208 | Lep d 2.0101 | Lepidoglyphus destructor.
| | Microorganisms, Molds, Yeast
| | m1 | Penicillium chrysogenum (Penicillium notatum) | Penicillium chrysogenum (Penicillium notatum).
| | m2 | Cladosporium herbarum (Hormodendrum) | Cladosporium herbarum (Hormodendrum).
| | m3 | Aspergillus fumigatus | Aspergillus fumigatus.
| | m4 | Mucor racemosus | Mucor racemosus.
| | m5 | Candida albicans | Candida albicans.
| | m7 | Botrytis cinerea | Botrytis cinerea.
| | m8 | Drechslera halodes (Setomelanomma rostrata, Helminthosporium halodes, Helminthosporium interseminatum) | Drechslera halodes (Setomelanomma rostrata, Helminthosporium halodes.
| | m9 | Fusarium moniliforme (Fusarium proliferatum) | Fusarium moniliforme (Fusarium proliferatum).
| | m10 | Stemphylium botryosum | Stemphylium herbarum (Stemphylium botryosum).
| | m11 | Rhizopus nigricans | Rhizopus nigricans.
| | m12 | Aureobasidium pullulans | Aureobasidium pullulans.
| | m13 | Phoma betae | Phoma betae.
| | m14 | Epicoccum purpurascens | Epicoccum purpurascens (Epicoccum nigrum).
| | m15 | Trichoderma viride | Trichoderma viride.
| | m16 | Curvularia lunata | Curvularia lunata, Curvularia specifera (K923044).
| | m17 | Cladosporium fulvum | Cladosporium fulvum.
| | m18 | Fusarium culmorum | Fusarium culmorum.
| | m19 | Aspergillus versicolor | Aspergillus versicolor.
| | m20 | Mucor mucedo | Mucor mucedo.
| | m21 | Aspergillus clavatus | Aspergillus clavatus.
| | m22 | Micropolyspora faeni | Saccharopolyspora rectivirgula (Micropolyspora faeni).
| | m23 | Thermoactinomyces vulgaris | Thermoactinomyces vulgaris.
| | m24 | Stachybotrys atra | Stachybotrys chartarum (Stachybotrys atra).
| | m24 | Paecilomyces spp | Paecilomyces spp.
| | m25 | Aspergillus versicolor | Aspergillus versicolor.
| | m25 | Penicillium brevicompactum | Penicillium brevicompactum.
| | m26 | Cladosporium cladosporioides | Cladosporium cladosporioides.
| | m26 | Penicillium citrinum | Penicillium citrinum.
| | m27 | Penicillium spp | Penicillium spp.
| | m29 | Aspergillus repens | Aspergillus repens.
| | m30 | Penicillium roqueforti | Penicillium roqueforti.
| | m32 | Cladosporium cladosporioides | Cladosporium cladosporioides.
| | m34 | Serpula lacrymans | Serpula lacrymans.
| | m36 | Aspergillus terreus | Aspergillus terreus.
| | m37 | Trichophyton mentagrophytes | Trichophyton mentagrophytes.
| | m40 | Aspergillus amstelodami | Aspergillus amstelodami.
| | m43 | Saccharomyces Carlsberg | Saccharomyces carlsbergensis.
| | m44 | Saccharomyces cerevisiae | Saccharomyces cerevisiae.
| | m45 | Hormodendrum hordei | Hormodendrum hordei.
| | m46 | Bipolaris spicifera | Bipolaris spicifera.
| | m47 | Aspergillus nidulans | Aspergillus nidulans.
| | m48 | Aspergillus oryzae | Aspergillus oryzae.
| | m49 | Fusarium oxysporum | Fusarium oxysporum.
| | m50 | Micropolyspora faeni | Saccharopolyspora rectivirgula (Micropolyspora faeni).
| | m51 | Thermoactinomyces vulgaris | Thermoactinomyces vulgaris.
| | m53 | Microspora canis | Microsporum canis (Microspora canis).
| | m54 | Aspergillus flavus | Aspergillus flavus.
| | m63 | Helminthosporium intersemin | Helminthosporium intersemin.
| | m66 | Mucor plumbeus | Mucor plumbeus.
| | m67 | Mycogone | Mycogone perniciosa.
| | m68 | Nigrospora oryzae | Nigrospora oryzae.
| | m69 | Rhodotorula | Rhodotorula rubra (Rhodotorula mucilaginosa).
| | m70 | Malassezia furfur (Pityrosporum orbiculare) | Malassezia furfur (Pityrosporum orbiculare).
| | m71 | Spondylocladium | Spondylocladium spp.
| | m72 | Epidermophyton | Epidermophyton floccosum.
| | m73 | Epicoccum nigrum | Epicoccum purpurascens (Epicoccum nigrum).
| | m80 | Staphylococcal enterotoxin A (Sta a SEA) | Staphylococcus aureus.
| | m80 | Helminthosporium spp | Helminthosporium spp.
| | m81 | Staphylococcal enterotoxin B (Sta a SEB) | Staphylococcus aureus.
| | m88 | Stemphylium solani | Stemphylium solani.
| | m93 | Gliocladium fimbriatum | Gliocladium fimbriatum.
| | m94 | Phycomyces blakesleeanus | Phycomyces blakesleeanus.
| | m201 | Tilletia tritici (Ustilago nuda, Ustilago tritici) (Barley smut) | Tilletia tritici (Ustilago nuda, Ustilago tritici).
| | m202 | Acremonium kiliense (Cephalosporium acremonium) | Acremonium kiliense (Cephalosporium acremonium).
| | m203 | Trichosporon pullulans | Trichosporon pullulans.
| | m204 | Ulocladium chartarum | Ulocladium chartarum.
| | m205 | Trichophyton rubrum | Trichophyton rubrum.
| | m207 | Aspergillus niger | Aspergillus niger.
| | m208 | Chaetomium globosum | Chaetomium globosum.
| | m209 | Penicillium frequentans | Penicillium glabrum (Penicillium frequentans).
| | m209 | Stachybotrys chartarum | Stachybotrys chartarum (Stachybotrys atra).
| | m210 | Trichophyton mentagrophytes var. goetzii | Trichophyton mentagrophytes var. goetzii.
| | m211 | Trichophyton mentagrophytes var. interdigitale | Trichophyton mentagrophytes var. interdigitale.
| | m211 | Oat smut | Ustilago avenae.
| | m212 | Micropolyspora faeni | Saccharopolyspora rectivirgula (Micropolyspora faeni).
| | m212 | Geotrichum candidum | Geotrichum candidum.
| | m213 | Bermuda grass smut | Ustilago cynodontis.
| | m214 | Johnson grass smut | Sphacelotheca cruenta.
| | m215 | Corn smut | Ustilago maydis.
| | m218 | Asp f 1.0101 | Aspergillus fumigatus.
| | a3050 | Asp r 1 | Aspergillus restrictus.
| | m219 | Asp f 2 | Aspergillus fumigatus.
| | m220 | Asp f 3.0101 | Aspergillus fumigatus.
| | m221 | Asp f 4 | Aspergillus fumigatus.
| | m222 | Asp f 6.0101 | Aspergillus fumigatus.
| | m223 | Staphylococcal enterotoxin C (Sta a SEC) | Staphylococcus aureus.
| | m224 | Staphylococcal enterotoxin D (Sta a SED) | Staphylococcus aureus.
| | m226 | Staphylococcal enterotoxin TSST (Sta a TSST) | Staphylococcus aureus.
| | m227 | Malassezia spp. (Pityrosporum spp.) | Malassezia spp. (Pityrosporum spp.).
| | m228 | Aspergillus flavus.
| | | m229 | Alt a 1.0101 | Alternaria alternata (Alternaria tenuis).
| | m230 | Alt a 6.0101 | Alternaria alternata (Alternaria tenuis).
| | m231 | Cla h 8.0101 | Cladosporium herbarum (Hormodendrum).
| | m300 | Eurotium spp | Eurotium spp.
| | m304 | Aspergillus oryzae | Aspergillus oryzae.
| | m305 | Penicillium brevicompactum | Penicillium brevicompactum.
| | m309 | Aspergillus terreus | Aspergillus terreus.
| | m310 | Aspergillus nidulans | Aspergillus nidulans.
| | m311 | Aspergillus flavus | Aspergillus flavus.
| | m312 | Aspergillus clavatus | Aspergillus clavatus.
| | Epidermal & Animal
| | e6 | Guinea pig epithelium | Cavia porcellus.
| | e7 | Pigeon droppings | Columba palumbus, Columba livia.
| | e25 | Chicken serum | Gallus domesticus (Gallus gallus domesticus; Gallus spp.).
| | e26 | Parrot serum | Psittacoidea spp.
| | e62 | Camel | Camelus dromedaries.
| | e70 | Goose feathers | Anser anser.
| | e71 | Mouse epithelium | Mus musculus (Mus spp.).
| | e73 | Rat epithelium | Rattus norvegicus.
| | e74 | Rat urine proteins | Rattus norvegicus, Rattus rattus.
| | e75 | Rat serum proteins | Rattus norvegicus, Rattus rattus.
| | e76 | Mouse serum proteins | Mus musculus (Mus spp.).
| | e77 | Budgerigar droppings | Melopsittacus undulatus.
| | e78 | Budgerigar feathers | Melopsittacus undulatus.
| | e79 | Budgerigar serum proteins | Melopsittacus undulatus.
| | e80 | Goat epithelium | Capra hircus.
| | e81 | Sheep epithelium | Ovis aries (Ovis spp.).
| | e82 | Rabbit epithelium | Oryctolagus cuniculus (Oryctolagus spp.).
| | e83 | Swine epithelium | Sus scrofa (Sus scrofa domesticus; Sus spp.).
| | e84 | Hamster epithelium | Cricetus cricetus, Mesocricetus auratus, and Phodopus sungorus.
| | e85 | Chicken feathers | Gallus domesticus (Gallus gallus domesticus; Gallus spp.).
| | e86 | Duck feathers | Anas platyrhynchos.
| | e87 | Rat epithelium, serum proteins, and urine proteins | Rattus norvegicus Rattus rattus.
| | e88 | Mouse epithelium, serum proteins, and urine proteins (mouse) | Mus musculus (Mus spp.).
| | e89 | Turkey feathers | Meleagris gallopavo.
| | e90 | Budgerigar serum proteins, feathers, and droppings | Melopsittacus undulatus.
| | e91 | Pigeon serum proteins, feathers, and droppings | Streptopelia roseogrisea, Psittacidae spp.
| | e92 | Parrot serum proteins, feathers, and droppings | Ara spp.
| | e93 | Pigeon serum proteins | Streptopelia roseogrisea.
| | e94 | Fel d 1.0101 | Felis domesticus.
| | a345 | Fel d 1 | Felis domesticus.
| | e98 | Parrot droppings | Psittacoidea spp.
| | e101 | Can f 1.0101 | Canis familiaris (Canis domesticus).
| | a174 | Can f 1 | Canis familiaris (Canis domesticus).
| | e102 | Can f 2.0101 | Canis familiaris (Canis domesticus).
| | e196 | Parakeet feathers | Nymphicus hollandicus.
| | e197 | Parakeet droppings | Nymphicus hollandicus.
| | e198 | Parakeet serum | Nymphicus hollandicus.
| | e199 | Canary bird serum | Serinus canarius.
| | e200 | Canary bird droppings | Serinus canarius.
| | e201 | Canary bird feathers (Canary feathers) | Serinus canarius.
| | e202 | Reindeer epithelium | Rangifer tarandus.
| | e203 | Mink epithelium | Mustela spp.
| | e204 | Bos d 6 | Bos domesticus (Bos taurus; Bos spp.).
| | e205 | Horse, serum proteins | Equus caballus (Equus spp.).
| | e206 | Rabbit, serum proteins | Oryctolagus cuniculus (Oryctolagus spp.).
| | e208 | Chinchilla epithelium | Chinchilla laniger.
| | e209 | Gerbil epithelium | Meriones unguiculatus.
| | e210 | Fox epithelium | Vulpes vulpes.
| | e211 | Rabbit, urine proteins | Oryctolagus cuniculus (Oryctolagus spp.).
| | e212 | Swine, urine proteins | Sus scrofa (Sus scrofa domesticus; Sus spp.).
| | e213 | Parrot feathers | Ara spp.
| | e214 | Finch feathers | Lonchura domestica.
| | e215 | Pigeon feathers | Streptopelia roseogrisea (Streptopelia spp.), Columbia spp.
| | e216 | Deer epithelium | Dama dama.
| | e217 | Ferret epithelium | Mustela putorius.
| | e218 | Chicken droppings | Gallus domesticus (Gallus gallus domesticus; Gallus spp.).
| | e219 | Chicken, serum proteins | Gallus domesticus (Gallus gallus domesticus; Gallus spp.).
| | e220 | Fel d 2, Cat serum albumin | Felis domesticus.
| | e221 | Can f 3 | Canis familiaris (Canis domesticus) (Dog serum albumin).
| | e222 | Swine serum albumin (Sus s PSA) | Sus scrofa (Sus scrofa domesticus; Sus spp.).
| | e225 | Lovebird feathers | Psittacoidea agapomis.
| | e226 | Can f 5.0101 | Canis familiaris.
| | e227 | Equ c 1.0101 | Equus caballus.
| | e228 | Fel d 4.0101 | Felis domesticus.
| | e230 | Equ c 3 | Equus caballus.
| | e231 | Mus m 1 | Mus musculus.
| | Food
| | f9 | Rice | Oryza sativa.
| | f12 | Pea (green pea) | Pisum sativum.
| | f15 | White bean | Phaseolus vulgaris.
| | f19 | Cayenne pepper | Capsicum frutescens (Capsicum annum).
| | f21 | Sugar cane | Saccharum officinarum.
| | f22 | Raspberry | Rubus idaeus.
| | f26 | Pork | Sus scrofa (Sus scrofa domesticus; Sus spp.).
| | f29 | Watermelon | Citrullus lanatus (Citrullus vulgaris).
| | f31 | Carrot | Daucus carota.
| | f32 | Oyster mushroom | Pleurotus ostreatus.
| | f33 | Orange | Citrus sinensis.
| | f35 | Potato | Solanum tuberosum.
| | f43 | Mother's milk | Homo sapiens.
| | f44 | Strawberry | Fragaria vesca (Fragaria spp.).
| | f45 | Yeast, baker's | Saccharomyces cerevisiae.
| | f46 | Pepper, Red | Capsicum annuum.
| | f47 | Garlic | Allium sativum.
| | f48 | Onion | Allium cepa.
| | f49 | Apple | Malus x domestica (Malus spp.).
| | f51 | Bamboo shoot | Phyllostachys pubescens.
| | f52 | Cacao/chocolate | Theobroma cacao.
| | f54 | Sweet potato | Ipomoea batatas.
| | f55 | Common millet | Panicum miliaceum.
| | f56 | Foxtail millet | Setaria italica.
| | f57 | Japanese millet | Echinochloa crus-galli.
| | f58 | Pacific squid | Todarodes pacificus.
| | f59 | Octopus | Octopus vulgaris (Octopus spp.).
| | f63 | Kefir | NA.
| | f67 | Parmesan cheese | NA.
| | f81 | Cheese, cheddar type | NA.
| | f82 | Cheese, mold type | NA.
| | f83 | Chicken | Gallus domesticus (Gallus gallus domesticus; Gallus spp.).
| | f86 | Parsley | Petroselinum crispum.
| | f87 | Melon | Cucumis melo Cucumis melo + Citrullus lanatus.
| | f88 | Mutton (lamb) | Ovis aries (Ovis spp.).
| | f90 | Malt | Hordeum vulgare.
| | f92 | Banana | Musa spp.
| | f93 | Cacao | Theobroma cacao.
| | f94 | Pear | Pyrus communis (Pyrus spp.).
| | f97 | Yam | Dioscorea spp. Dioscorea opposita.
| | f97 | Chamomile tea | Matricaria chamomilla.
| | f98 | Gliadin | Triticum aestivum (Triticum spp.).
| | f102 | Cantaloupe | Cucumis melo var. cantalupensis.
| | f105 | Chocolate | Theobroma cacao.
| | f109 | Cottonseed | Gossypium hirsutum.
| | f110 | Giant radish | Raphanus sativus.
| | f118 | Zucchini | Cucurbita pepo.
| | f119 | Radish | Raphanus sativus.
| | f120 | Venison | Capreolus capeolus.
| | f121 | Pinto bean | Phaseolus vulgaris.
| | f122 | Cheese, American | NA.
| | f127 | Black-eyed pea | Vigna unguiculata.
| | f131 | Black Olive | Olea europaea.
| | f136 | Red beet | Beta vulgaris var. conditiva.
| | f139 | Goat's Cheese | Capra aegagrus.
| | f140 | Bran | NA.
| | f141 | Corn (vegetables) | Zea mays.
| | f152 | Green bell pepper | Capsicum annuum.
| | f155 | Brewer's yeast | Saccharomyces carlsbergensis.
| | f157 | Duck | Anas domesticus.
| | f158 | Goose | Anser anser.
| | f160 | Camembert cheese | NA.
| | f162 | Nectarine | Prunus persica var. nucipersica.
| | f163 | Kohlrabi | Brassica oleracea var. gongylodes.
| | f65 | Perch
| | | f166 | Leek | Allium porrum.
| | f170 | Cheese (Switzerland) (Swiss cheese) | NA.
| | f174 | Fig | Ficus carica.
| | f177 | Cranberry | Vaccinium macrocarpon.
| | f179 | Raisin | Vitis spp.
| | f182 | Lima bean | Phaseolus lunatus.
| | f198 | Flaxseed (bruised grain) | Linum usitatissimum.
| | f199 | Untreated native milk | Bos domesticus (Bos taurus; Bos spp.).
| | f208 | Lemon | Citrus limon.
| | f209 | Grapefruit | Citrus paradisi.
| | f210 | Pineapple | Ananas comosus.
| | f211 | Blackberry | Rubus fruticosus.
| | f212 | Mushroom (champignon) | Agaricus hortensis (Agaricus spp.).
| | f213 | Rabbit | Oryctolagus cuniculus (Oryctolagus spp.).
| | f214 | Spinach | Spinacia oleracea.
| | f215 | Lettuce | Lactuca sativa.
| | f216 | Cabbage | Brassica oleracea var. capitata.
| | f217 | Brussels sprouts | Brassica oleracea var. gem.
| | f218 | Paprika, sweet pepper | Capsicum annuum.
| | f219 | Fennel seed | Foeniculum vulgare.
| | f219 | Sage | Salvia officinalis.
| | f220 | Cinnamon | Cinnamomum spp.
| | f221 | Coffee | Coffea spp.
| | f222 | Tea | Camellia sinensis.
| | f223 | Green olive | Olea europaea.
| | f225 | Summer squash, pumpkin | Cucurbita pepo.
| | f225 | Pumpkin | Cucurbita maxima.
| | f226 | Pumpkin seed | Cucurbita pepo.
| | f227 | Sugar-beet seed | Beta vulgaris.
| | f229 | Safflower Seed | Carthamus tinctorius.
| | f231 | Milk, boiled | Bos domesticus (Bos taurus; Bos spp.).
| | f234 | Vanilla | Vanilla planifolia.
| | f237 | Apricot | Prunus armeniaca.
| | f241 | Gouda cheese | NA.
| | f242 | Cherry | Prunus avium.
| | f244 | Cucumber | Cucumis sativus.
| | f246 | Guar, guar gum | Cyamopsis tetragonoloba.
| | f247 | Honey | NA.
| | f248 | Rosemary | Rosmarinus officinalis.
| | f254 | Plaice | Pleuronectes platessa.
| | f255 | Plum | Prunus domestica, Prunus americana.
| | f258 | Squid | Loligo spp.
| | f259 | Grape | Vitis vinifera (Vitis spp.).
| | f260 | Broccoli | Brassica oleracea var. italica (Brassica oleracea var. cultivar).
| | f261 | Asparagus | Asparagus officinalis.
| | f262 | Aubergine, eggplant | Solanum melongena.
| | f263 | Green pepper | Piper nigrum, Capsicum annuum.
| | f264 | Eel | Anguilla anguilla.
| | f265 | Caraway | Carum carvi.
| | f265 | Cumin | Cuminum cyminum.
| | f266 | Mace | Myristica fragrans.
| | f267 | Cardamon | Elettaria cardamomum.
| | f268 | Clove | Syzygium aromaticum.
| | f269 | Basil | Ocimum basilicum.
| | f270 | Ginger | Zingiber officinale.
| | f271 | Anise | Pimpinella anisum.
| | f272 | Tarragon | Artemisia dracunculus.
| | f273 | Thyme | Thymus vulgaris.
| | f274 | Marjoram | Origanum majorana.
| | f275 | Lovage | Levisticum officinale.
| | f276 | Fennel, fresh | Foeniculum vulgare.
| | f277 | Dill | Anethum graveolens.
| | f278 | Bay leaf | Laurus nobilis.
| | f279 | Chili pepper | Capsicum frutescens.
| | f280 | Black pepper | Piper nigrum.
| | f281 | Curry (Santa Maria) | NA.
| | f282 | Nutmeg | Myristica fragrans.
| | f283 | Oregano | Origanum vulgare.
| | f284 | Turkey meat | Meleagris gallopavo.
| | f285 | Elk/moose meat | Alces spp.
| | f286 | Mare's milk | Equus caballus (Equus spp.).
| | f287 | Red kidney bean | Phaseolus vulgaris.
| | f288 | Blueberry | Vaccinium myrtillus (Vaccinium spp.).
| | f289 | Date | Phoenix dactylifera.
| | f291 | Cauliflower | Brassica oleracea var. botrytis.
| | f292 | Guava | Psidium guajava.
| | f293 | Papaya | Carica papaya.
| | f294 | Passion fruit, Maracuja | Passiflora edulis (Passiflora spp.).
| | f295 | Carambola | Averrhoa carambola.
| | f296 | Carob | Ceratonia siliqua.
| | f297 | Gum Arabic | Acacia senegal (Acacia spp.).
| | f298 | Tragacanth | Astragalus spp.
| | f299 | Sweet chestnut (chestnut) | Castanea sativa.
| | f300 | Pinto bean | Phaseolus spp.
| | f301 | Persimmon (kaki fruit, sharon) | Diospyros kaki.
| | f302 | Mandarin (tangerine, clementine, satsumas) | Citrus reticulata.
| | f305 | Fenugreek | Trigonella foenum-graecum.
| | f306 | Lime | Citrus aurantifolia.
| | f307 | Hake | Merluccius merluccius.
| | f308 | Sardine (pilchard) | Sardina pilchardus.
| | f310 | Blue vetch | Lathyrus sativus.
| | f311 | Megrim | Lepidorhombus whiffiagonis.
| | f315 | Green bean | Phaseolus vulgaris.
| | f316 | Rape seed | Brassica napus.
| | f317 | Coriander | Coriandrum sativum.
| | f318 | Jack fruit | Artocarpus heterophyllus.
| | f319 | Beetroot | Beta vulgaris.
| | f320 | Crayfish | Astacus astacus.
| | f321 | Horse meat | Equus caballus (Equus spp.).
| | f322 | Red currant | Ribes sylvestre.
| | f324 | Hop (fruit cone) | Humulus lupulus.
| | f325 | Saffron | Colchicum autumnale.
| | f328 | Fig | Ficus carica.
| | f329 | Watermelon | Citrullus lanatus.
| | f330 | Rose hip | Rosa spp.
| | f331 | Saffron | Crocus sativus.
| | f332 | Mint | Mentha piperita.
| | f333 | Linseed | Linum usitatissimum.
| | f336 | Jujube | Ziziphus jujuba.
| | f336 | Wine vinegar | Vitis vinifera (Vitis spp.).
| | f337 | Sole | Solea solea.
| | f337 | English sole | Parophrys vetulus.
| | f338 | Wine, white | Vitis vinifera (Vitis spp.).
| | f339 | Allspice | Pimenta dioica.
| | f339 | Wine, red | Vitis vinifera (Vitis spp.).
| | f341 | Cranberry | Vaccinium oxycoccus, Vaccinium macrocarpon.
| | f342 | Olive (black, fresh) | Olea europaea.
| | f343 | Raspberry | Rubus idaeus.
| | f344 | Sage | Salvia officinalis.
| | f346 | Chives | Allium schoenoprasum.
| | f347 | Quinoa | Chenopodium quinoa.
| | f348 | Litchi | Litchi chinensis.
| | f349 | Chum salmon roe | Oncorhynchus keta.
| | f358 | Artichoke | Cynara scolymus.
| | f360 | Yogurt | NA.
| | f368 | Black bass | Micropterus dolomieu (Micropterus dolomieui).
| | f374 | Karaya gum | Sterculia urens.
| | f375 | Horseradish | Armoracia rusticana.
| | f377 | Maple syrup | NA.
| | f379 | Okra | Abelmoschus esculentus.
| | f382 | Beet, sugar | Beta vulgaris var. altissima.
| | f401 | Loquat | Eriobotrya japonica.
| | f402 | Fig | Ficus carica.
| | f403 | Brewer's yeast | Saccharomyces cerevisiae.
| | f405 | Mint | Mentha spp.
| | f406 | Arugula | Eruca vesicaria.
| | House Dust
| | h1 | Greer Labs., Inc | NA.
| | h2 | Hollister-Stier Labs | NA.
| | h6 | Japan | NA.
| | Venoms & Insects
| | i7 | Midge | Chironomus yoshimatsui.
| | i8 | Moth | Bombyx mori, Heterocera spp.
| | i47 | Water flea | Daphnia spp.
| | i49 | Deer fly | Chrysops spp.
| | i51 | Black ant | Camponotus pennsylvanicus.
| | i54 | Flea mix (dog/cat), common flea | Ctenocephalides spp.
| | i71 | Mosquito | Aedes communis, Aedes spp. and Culex spp.
| | i72 | Green nimitti | Cladotanytarsus lewisi.
| | i73 | Blood worm | Chironomus thummi, Chironomusri parius, Chironomus spp.
| | i75 | European hornet | Vespa crabro.
| | i76 | Berlin beetle | Trogoderma angustum.
| | i77 | European paper wasp | Polistes dominulus.
| | i78 | Fly | Musca domestica.
| | i80 | Bumblebee | Bombus pennsylvanicus.
| | i201 | Horse bot fly | Gasterophilus intestinalis.
| | i202 | Grain weevil | Sitophilus granarius.
| | i203 | Mediterranean flour moth | Ephestia kuehniella (Anagasta kuehniella).
| | i204 | Horse fly | Tabanus spp.
| | i205 | Bumblebee | Bombus terrestris.
| | i208 | Api m 1.0101 | Apis mellifera.
| | a45 | Api m 1 | Apis mellifera.
| | i209 | Ves v 5.0101 | Vespula vulgaris.
| | a670 | Ves v 5 | Vespula vulgaris.
| | i210 | Pol d 5.0101 | Polistes dominulus.
| | i211 | Ves v 1.0101 | Vespula vulgaris.
| | i213 | Api m 4 | Apis mellifera.
| | i214 | Api m 2 | Apis mellifera.
| | i215 | Api m 3 | Apis mellifera.
| | i216 | Api m 5 | Apis mellifera.
| | i217 | Api m 10 | Apis mellifera.
| | i220 | Bla g 1.0101 | Blattella germanica.
| | i221 | Bla g 2.0101 | Blattella germanica.
| | i222 | Bla g 5.0101 | Blattella germanica.
| | i223 | Bla g 7 | Blattella germanica.
| | a46 | Api m 2 | Apis mellifera.
| | Miscellaneous
| | o1 | Cotton, crude fibers | Gossypium spp.
| | o3 | Cotton (treated) | Gossypium spp.
| | o70 | Seminal fluid | Homo sapiens.
| | o71 | Staphylococcus aureus | Staphylococcus aureus.
| | o72 | Pichia pastoris crude extract customer specific | Pichia pastoris.
| | o72 | Sperm-sediment | Homo sapiens.
| | o73 | Pichia pastoris crude extr. vector customer specific | Pichia pastoris.
| | o74 | Pichia pastoris with vector customer specific | Pichia pastoris.
| | o201 | Tobacco leaf, tobacco dust | Nicotiana tabacum.
| | o202 | Artemia salina, fish feed | Artemia salina.
| | o203 | Tetramin, fish feed | NA.
| | o207 | Daphnia, fish feed | Daphnia spp.
| | o211 | Mealworm | Tenebrio molitor.
| | o212 | Streptavidin | Streptomyces avidini.
| | o213 | MBP (maltose binding protein) | Escherichia coli.
| | o214 | CCD; MUXF3 from bromelain | Ananas comosus.
| | o72 | Enterotoxin A (Sta a SEA) | Staphylococcus aureus.
| | o73 | Enterotoxin B (Sta a SEB) | Staphylococcus aureus.
| | Parasites
| | p1 | Ascaris | Ascaris suum.
| | p2 | Echinococcus | Echinococcus granulosus.
| | p3 | Schistosoma | Schistosoma mansoni.
| | p4 | Anisakis (Herring Worm) | Anisakis simplex (Anisakis spp.).
| | p5 | Toxocara canis | Toxocara canis.
| | p10 | Ani s 3.0101 | Anisakis simplex (Anisakis spp.).
| | p11 | Ani s 1 | Anisakis simplex (Anisakis spp.).
| | Occupational
| | k4 | Threshing dust | NA.
| | k5 | Flax | NA.
| | k7 | Hay Dust | NA.
| | k8 | Hop (hops) | Humulus lupulus.
| | k12 | Grain mill dust | NA.
| | k14 | Kapok | NA.
| | k20 | Sheep's wool (treated) (wool) | Ovis aries (Ovis spp.).
| | k21 | Sheep's wool (Untreated) | Ovis aries (Ovis spp.).
| | k23 | Straw Dust | NA.
| | k33 | Oak | NA.
| | k70 | Green coffee bean | Coffea spp.
| | k71 | Castor bean | Ricinus communis.
| | k72 | Ispaghula | Plantago psyllium/Plantago ovata.
| | k73 | Silk waste | NA.
| | k74 | Silk | Bombyx mori.
| | k75 | Isocyanate TDI (Toluene diisocyanate) | NA.
| | k76 | Isocyanate MDI (Diphenylmethane diisocyanate) | NA.
| | k77 | Isocyanate HDI (Hexamethylen diisocyanate) | NA.
| | k78 | Ethylene oxide | NA.
| | k79 | Phthalic anhydride | NA.
| | k80 | Formaldehyde/Formalin | NA.
| | k81 | Ficus | Ficus benjamina (Ficus spp.).
| | k83 | Cotton seed | Gossypium hirsutum.
| | k84 | Sunflower seed | Helianthus annuus.
| | k85 | Chloramin T | NA.
| | k86 | Trimellitic anhydride, TMA | NA.
| | k87 | Asp o 21, alpha-amylase | Aspergillus oryzae.
| | k89 | Orris root | Iris florentina.
| | k99 | HSA (Human Serum Albumin) (Hom s HSA) | Homo sapiens.
| | k201 | Car p 1, Papain | Carica papaya.
| | k202 | Ana c 2, Bromelain | Ananas comosus.
| | k204 | Maxatase | Bacillus licheniformis.
| | k205 | Alcalase | Bacillus spp.
| | k206 | Savinase, Protease 1 (Bac l Subtilisin) | Bacillus spp.
| | k208 | Gal d 4, Lysozyme | Gallus domesticus (Gallus gallus domesticus; Gallus spp.).
| | k209 | Hexahydrophtalic anhydrid | NA.
| | k210 | Maleic anhydride | NA.
| | k211 | Methyltetrahydrophtalic anhydrid | NA.
| | k212 | Abachi wood dust | Triplochiton scleroxylon.
| | k213 | Pepsin (Sus s Pepsin) | Sus scrofa (Sus scrofa domesticus; Sus spp.).
| | k213 | TCPA | NA.
| | k214 | Bougainvillea | Bougainvillea spp.
| | k225 | Horse radish peroxidase (Arm r HRP) | Armoracia rusticana.
| | k226 | Ascorbate oxidase (Cuc p ascorbate oxidase) | Cucurbita pepo.
| | k301 | Flour dust | Triticum spp.
| | k501 | Savinase customer specific | Proprietary knowledge of customer.
| | k502 | Lipolase customer specific | Proprietary knowledge of customer.
| | k503 | Termamyl customer specific | Proprietary knowledge of customer.
| | k504 | Clazinase customer specific | Proprietary knowledge of customer. |
[47 FR 50823, Nov. 9, 1982, as amended at 84 FR 71800, Dec. 30, 2019]
|
|
|
Sec. 866.5760 Tryptase test system.
|
|
(a) Identification. A tryptase test system is a device that aids in the diagnosis of systemic mastocytosis. It is intended for in vitro diagnostic use as an aid in the clinical diagnosis of patients with a suspicion of systemic mastocytosis in conjunction with other clinical and laboratory findings.
(b) Classification. Class II (special controls). The special control is FDA's guideline entitled "Class II Special Controls Guideline: Tryptase Test System as an Aid in the Diagnosis of Systemic Mastocytosis." For availability of the document, see § 866.1(e).
[79 FR 56010, Sept. 18, 2014]
|
|
|
Sec. 866.5765 Retinol-binding protein immunological test system.
|
|
(a) Identification. A retinol-binding protein immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the retinol-binding protein that binds and transports vitamin A in serum and urine. Measurement of this protein may aid in the diagnosis of kidney disease and in monitoring patients with kidney transplants.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 65 FR 2313, Jan. 14, 2000]
|
|
|
Sec. 866.5775 Rheumatoid factor immunological test system.
|
|
(a) Identification. A rheumatoid factor immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the rheumatoid factor (antibodies to immunoglobulins) in serum, other body fluids, and tissues. Measurement of rheumatoid factor may aid in the diagnosis of rheumatoid arthritis.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5785 Anti-Saccharomyces cerevisiae (S. cerevisiae) antibody (ASCA) test systems.
|
|
(a) Identification. The Anti-Saccharomyces cerevisiae (S. cerevisiae ) antibody (ASCA) test system is an in vitro diagnostic device that consists of the reagents used to measure, by immunochemical techniques, antibodies to S. cerevisiae (baker's or brewer's yeast) in human serum or plasma. Detection of S. cerevisiae antibodies may aid in the diagnosis of Crohn's disease.
(b) Classification. Class II (special controls). The special control is FDA's "Guidance for Industry and FDA Reviewers: Class II Special Control Guidance Document for Anti-Saccharomyces cerevisiae (S. cerevisiae ) Antibody (ASCA) Premarket Notifications."
[65 FR 70307, Nov. 22, 2000]
|
|
|
Sec. 866.5800 Seminal fluid (sperm) immunological test system.
|
|
(a) Identification. A seminal fluid (sperm) immunological test system is a device that consists of the reagents used for legal purposes to identify and differentiate animal and human semen. The test results may be used as court evidence in alleged instances of rape and other sex-related crimes.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[54 FR 25047, June 12, 1989, as amended at 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5820 Systemic lupus erythematosus immunological test system.
|
|
(a) Identification. A systemic lupus erythematosus (SLE) immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the autoimmune antibodies in serum and other body fluids that react with cellular nuclear double-stranded deoxyribonucleic acid (DNA) or other nuclear constituents that are specifically diagnostic of SLE. Measurement of nuclear double-stranded DNA antibodies aids in the diagnosis of SLE (a multisystem autoimmune disease in which tissues are attacked by the person's own antibodies).
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5830 Brain trauma assessment test.
|
|
(a) Identification. A brain trauma assessment test is a device that consists of reagents used to detect and measure brain injury biomarkers in human specimens. The measurements aid in the evaluation of patients with suspected mild traumatic brain injury in conjunction with other clinical information to assist in determining the need for head imaging per current standard of care.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) The 21 CFR 809.10(b) compliant labeling must include detailed descriptions of and results from performance testing conducted to evaluate precision, accuracy, linearity, analytical sensitivity, interference, and cross-reactivity. This information must include the following:
(i) Performance testing of device precision must, at minimum, use one unmodified clinical specimen from the intended use population with concentration of the brain injury biomarker(s) near the medical decision point. Contrived specimens that have been generated from pooling of multiple samples or spiking of purified analyte to cover the measuring range may be used, but the contrived samples must be prepared to mimic clinical specimens as closely as possible. This testing must evaluate repeatability and reproducibility using a protocol from an FDA-recognized standard.
(ii) Device performance data must be demonstrated through a clinical study and must include the following:
(A) Data demonstrating clinical validity including the clinical sensitivity and specificity, and positive and negative predictive value of the test in the intended use population of patients with suspected mild traumatic brain injury (i.e., Glasgow Coma Score (GCS) of 13-15), or equivalent standard of care for determination of severity of traumatic brain injury (TBI).
(B) Study must be performed using the operators and in settings that are representative of the types of operators and settings for which the device is intended to be used.
(C) All eligible subjects must meet the well-defined study inclusion and exclusion criteria that define the intended use population. The prevalence of diseased or injured subjects in the study population must reflect the prevalence of the device's intended use population, or alternatively, statistical measures must be used to account for any bias due to enrichment of subpopulations of the intended use population.
(D) All eligible subjects must have undergone a head computerized tomography (CT) scan or other appropriate clinical diagnostic standard used to determine the presence of an intracranial lesion as part of standard of care and must also be evaluated by the subject device. All clinical diagnostic standards used in the clinical study must follow standard clinical practice in the United States.
(E) Relevant demographic variables and baseline characteristics including medical history and neurological history. In addition, head injury characteristics, neurological assessments, and physical evidence of trauma must be provided for each subject. This information includes but is not limited to the following: Time since head injury, time from head injury to CT scan, time from head injury to blood draw, GCS score or equivalent, experience of loss of consciousness, presence of confusion, episodes of vomiting, post-traumatic amnesia characteristics, presence of post-traumatic seizures, drug or alcohol intoxication, mechanism of injury, acute intracranial lesion type, neurosurgical lesion, and cranial fracture.
(F) Each CT scan or other imaging result must be independently evaluated in a blinded manner by at least two board-certified radiologists to determine whether it is positive or negative as defined by the presence or absence of acute intracranial lesions. This independent review must be conducted without access to test results of the device. Prior to conducting the review, the criteria and procedures to be followed for scoring the images must be established, including the mechanism for determining consensus.
(G) All the clinical samples must be tested with the subject device blinded to the TBI status and the neurological-lesion-status of the subject.
(H) Details on how missing values in data are handled must be provided.
(I) For banked clinical samples, details on storage conditions and storage period must be provided. In addition, a specimen stability study must be conducted for the duration of storage to demonstrate integrity of archived clinical samples. The samples evaluated in the assay test development must not be used to establish the clinical validity of the assays.
(iii) Performance testing of device analytical specificity must include the most commonly reported concomitant medications present in specimens from the intended use population. Additionally, potential cross-reacting endogenous analytes must be evaluated at the highest concentration reported in specimens from the intended use population.
(iv) Expected/reference values generated by testing a statistically appropriate number of samples from apparently healthy normal individuals.
(2) The 21 CFR 809.10(a) and (b) compliant labeling must include the following limitations:
(i) A limiting statement that this device is not intended to be used a stand-alone device but as an adjunct to other clinical information to aid in the evaluation of patients who are being considered for standard of care neuroimaging.
(ii) A limiting statement that reads "A negative result is generally associated with the absence of acute intracranial lesions. An appropriate neuroimaging method is required for diagnosis of acute intracranial lesions."
(iii) As applicable, a limiting statement that reads "This device is for use by laboratory professionals in a clinical laboratory setting."
[83 FR 27701, June 14, 2018]
|
|
|
Sec. 866.5860 Total spinal fluid immunological test system.
|
|
(a) Identification. A total spinal fluid immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the total protein in cerebrospinal fluid. Measurement of spinal fluid proteins may aid in the diagnosis of multiple sclerosis and other diseases of the nervous system.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 61 FR 1119, Jan. 16, 1996; 66 FR 38793, July 25, 2001]
|
|
|
Sec. 866.5870 Thyroid autoantibody immunological test system.
|
|
(a) Identification. A thyroid autoantibody immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the thyroid autoantibodies (antibodies produced against the body's own tissues). Measurement of thyroid autoantibodies may aid in the diagnosis of certain thyroid disorders, such as Hashimoto's disease (chronic lymphocytic thyroiditis), nontoxic goiter (enlargement of thyroid gland), Grave's disease (enlargement of the thyroid gland with protrusion of the eyeballs), and cancer of the thyroid.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5880 Transferrin immunological test system.
|
|
(a) Identification. A transferrin immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the transferrin (an iron-binding and transporting serum protein) in serum, plasma, and other body fluids. Measurement of transferrin levels aids in the diagnosis of malnutrition, acute inflammation, infection, and red blood cell disorders, such as iron deficiency anemia.
(b) Classification. Class II (performance standards).
|
|
|
Sec. 866.5890 Inter-alpha trypsin inhibitor immunological test system.
|
|
(a) Identification. An inter-alpha trypsin inhibitor immunological test system is a device that consists of the reagents used to measure by immunochemical techniques the inter-alpha trypsin inhibitor (a protein) in serum and other body fluids. Measurement of inter-alpha trypsin inhibitor may aid in the diagnosis of acute bacterial infection and inflammation.
(b) Classification. Class I (general controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to § 866.9.
[47 FR 50823, Nov. 9, 1982, as amended at 53 FR 11253, Apr. 6, 1988; 65 FR 2313, Jan. 14, 2000]
|
|
|
Sec. 866.5900 Cystic fibrosis transmembrane conductance regulator (CFTR) gene mutation detection system.
|
|
(a) Identification. The CFTR gene mutation detection system is a device used to simultaneously detect and identify a panel of mutations and variants in the CFTR gene. It is intended as an aid in confirmatory diagnostic testing of individuals with suspected cystic fibrosis (CF), carrier identification, and newborn screening. This device is not intended for stand-alone diagnostic purposes, prenatal diagnostic, pre-implantation, or population screening.
(b) Classification. Class II (special controls). The special control is FDA's guidance document entitled "Class II Special Controls Guidance Document: CFTR Gene Mutation Detection System." See § 866.1(e) for the availability of this guidance document.
[70 FR 61738, Oct. 26, 2005]
|
|
|
Sec. 866.5910 Quality control material for cystic fibrosis nucleic acid assays.
|
|
(a) Identification. Quality control material for cystic fibrosis nucleic acid assays. A quality control material for cystic fibrosis nucleic acid assays is a device intended to help monitor reliability of a test system by detecting analytical deviations such as those that may arise from reagent or instrument variation in genetic testing. This type of device includes recombinant, synthetic, and cell line-based DNA controls.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9. The special control is FDA's guidance document entitled "Class II Special Controls Guidance Document: Quality Control Material for Cystic Fibrosis Nucleic Acid Assays." See § 866.1(e) for the availability of this guidance document.
[72 FR 1176, Jan. 10, 2007, as amended at 84 FR 71811, Dec. 30, 2019]
|
|
|
Sec. 866.5930 Newborn screening test for severe combined immunodeficiency disorder (SCID).
|
|
(a) Identification. A newborn screening test for SCID is a prescription device intended to measure T-cell receptor excision circle (TREC) DNA obtained from dried blood spot specimens on filter paper using a polymerase chain reaction based test as an aid in screening newborns for SCID. Presumptive positive results must be followed up by diagnostic confirmatory testing. This test is not intended for use as a diagnostic test, or for screening of SCID-like syndromes, such as DiGeorge syndrome or Omenn syndrome. It is also not intended to screen for less acute SCID syndromes, such as leaky SCID or variant SCID.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) Premarket notification submissions must include the following information:
(i) The intended use must indicate:
(A) The test is not intended for diagnostic use, or for screening of SCID-like syndromes, such as DiGeorge syndrome or Omenn syndrome; and
(B) The test is not intended to screen for less acute SCID syndromes, such as leaky SCID or variant SCID.
(ii) A detailed description of all components in the test that includes:
(A) A detailed description of the test components, all required reagents, instrumentation and equipment, including illustrations or photographs of nonstandard equipment or methods;
(B) Detailed documentation of the device software including, but not limited to, standalone software applications and hardware-based devices that incorporate software;
(C) Specifications for the filter paper, which must be appropriately labeled for in vitro diagnostic use, to be used in specimen collection and how it will be used in specimen collection validation. These specifications must include: descriptive characteristics of the filter paper, instructions on how a lab should choose the appropriate filter paper, chemical properties of the filter paper, interference concerns associated with the chemicals in the filter paper, absorption properties of the filter paper, punch size, absorption capacity, testing for homogeneity of punches, diameter of the circle for the dried blood spot aliquot, absorption time, physical composition, and number and size of punches to be tested;
(D) Methodology and protocols for detection of T-cell receptor excision circles and methods for determination of results. The cutoff must be selected before conducting clinical and analytical studies;
(E) A description of the result outputs along with sample reports. Sample reports must include the scale used in reporting of results (e.g., TREC copies/[micro]L) and the range of values that will be reported out; and
(F) A description of appropriate internal and external controls that are recommended or provided. The description must identify those control elements that are incorporated into the testing procedure.
(iii) Information that demonstrates the performance characteristics of the test, including:
(A) Data that demonstrates the clinical validity of the device, using well characterized prospectively or retrospectively obtained clinical specimens representative of the intended use population. A minimum of 10 to 15 confirmed positive specimens must be obtained from more than 1 site, including relevant annotation, and, at 1 year or beyond, a SCID diagnosis by flow cytometry or clinically meaningful information regarding the status of the subject must be obtained. Additional specimens should have been obtained that are characterized by other disorders that can be found by screening specimens that have low or absent TREC (e.g., other T-cell lymphopenic disorders) to supplement the range of results. The clinical validation study must have a pre-specified clinical decision point (i.e., cutoff to distinguish positive and negative results). Results must be summarized in tabular format comparing interpretation of results to the reference method. Point estimates together with two-sided 95 percent confidence intervals must be provided for the positive percent agreement, negative percent agreement, and overall percent agreement. Data must include the retest rate, the false positive rate before retest, the final false positive rate, and the false negative rate;
(B) Device reproducibility data generated, using a minimum of three sites of which at least two must be external sites, with two operators at each site. Each site must conduct a minimum of five runs per operator over five nonconsecutive days evaluating a minimum of six different relevant TREC concentrations that span and are well distributed over the measuring range and include the clinical cutoff. Specimens must include cord blood and cord blood diluted with ABO matched adult blood specimens. Identical specimens from the same sample panel must be tested at each site. Each specimen must be run in triplicate and include controls run in triplicate. Results must be reported as the standard deviation and percentage coefficient of variation for each level tested. Results must also be displayed as a dichotomous variable around the cutoff. Total variation must be partitioned into the sum of within-lab and between-lab variations with pre-specified acceptance criteria and 95 percent confidence intervals for all data. Pre-specified acceptance criteria must be provided and followed;
(C) Device precision data using clinical samples to evaluate the within-lot, between-lot, within-run, between run, and total variation. A range of TREC levels of the specimen must include samples within the measuring range, samples above and below the measuring range, as well as with samples very near above and below the cutoff value. At least three replicates of each specimen must be tested with controls and calibrator(s) according to the device instructions for use. The precision study must use well characterized samples using different lots, instruments, and operators. Results must be summarized in tabular format. Pre-specified acceptance criteria must be provided and followed;
(D) Linearity of the test must be demonstrated using a dilution panel from clinical samples. The range of dilution samples must include samples within the measuring range, samples above and below the measuring range, as well as with samples very near above and below the cutoff value. Results of the regression analysis must be summarized in tabular format and fitted into a linear regression model with the individual measurement results against the dilution factors. Pre-specified acceptance criteria must be provided and followed;
(E) Device analytic sensitivity data, including limit of blank, limit of detection, and limit of quantification;
(F) Device specificity data, including interference, carryover, cross-contamination, and in silico analysis of potential off-target genomic sequences;
(G) Device stability data, including real-time stability of samples under various storage times, temperatures, and freeze-thaw conditions. A separate shipping stability study must be performed;
(H) Lot-to-lot reproducibility study of each filter paper that will be validated with the test. The lot-to-lot study must include a minimum of three lots of each blood spot card that will be validated with the test and be conducted over five nonconsecutive days. The sample panel must consist of specimens with a range of TREC levels and include samples within the measuring range, samples above and below the measuring range, and samples very near above and below the cutoff value. Multiple punches must be obtained from each card for demonstration of homogeneity of the analyte across the dried blood spot. Comparability of the test performance for each filter paper must be demonstrated. Stability and storage of TREC DNA on each blood spot card must be demonstrated. Results of the lot-to-lot study must be summarized providing the mean, standard deviation, and percentage coefficient of variation in a tabular format. Data must be calculated for within-run, between-run, within-lot, and between-lot. Data demonstrating the concordance between results across different filter papers must be provided. Study acceptance criteria must be provided and followed; and
(I) If applicable, a thermocycler reproducibility study must be performed using thermocyclers from three independent thermocyler manufacturers. The sample panel must consist of specimens with a range of TREC levels and must include samples within the measuring range, samples above and below the measuring range, and samples very near above and below the cutoff value. The study must be done using three filter paper lots and conducted over five nonconsecutive days. Results of the thermocycler reproducibility study must be summarized providing the mean, standard deviation, and percentage coefficient of variance in a tabular format. Data must be calculated for the within-run, between-run, within-lot, between-lot, and between thermocycler manufacturer study results. Study acceptance criteria must be provided and followed.
(iv) Identification of risk mitigation elements used by your device, including a description of all additional procedures, methods, and practices incorporated into the directions for use that mitigate risks associated with testing.
(2) Your § 809.10 compliant labeling must include:
(i) A warning statement that reads "This test is not intended for diagnostic use, preimplantation or prenatal testing, or for screening of SCID-like syndromes, such as DiGeorge syndrome or Omenn syndrome. It is also not intended to screen for less acute SCID syndromes, such as leaky SCID or variant SCID.";
(ii) A warning statement that reads "Test results are intended to be used in conjunction with other clinical and diagnostic findings, consistent with professional standards of practice, including confirmation by alternative methods and clinical evaluation, as appropriate.";
(iii) A description of the performance studies listed in paragraph (b)(1)(iii) and a summary of the results; and
(iv) A description of the filter paper specifications required for the test.
[82 FR 50079, Oct. 30, 2017]
|
|
|
Sec. 866.5940 Autosomal recessive carrier screening gene mutation detection system.
|
|
(a) Identification. Autosomal recessive carrier screening gene mutation detection system is a qualitative in vitro molecular diagnostic system used for genotyping of clinically relevant variants in genomic DNA isolated from human specimens intended for prescription use or over-the-counter use. The device is intended for autosomal recessive disease carrier screening in adults of reproductive age. The device is not intended for copy number variation, cytogenetic, or biochemical testing.
(b) Classification. Class II (special controls). The device is exempt from the premarket notification procedures in subpart E of part 807 of this chapter subject to the limitations in § 866.9, except § 866.9(c)(2). Autosomal recessive carrier screening gene mutation detection system must comply with the following special controls:
(1) If the device is offered over-the-counter, the device manufacturer must provide information to a potential purchaser or actual test report recipient about how to obtain access to a board-certified clinical molecular geneticist or equivalent to assist in pre- and post-test counseling.
(2) The device must use a collection device that is FDA cleared, approved, or classified as 510(k) exempt, with an indication for in vitro diagnostic use in DNA testing.
(3) The device's labeling must include a prominent hyperlink to the manufacturer's public Web site where the manufacturer shall make the information identified in this section publicly available. The manufacturer's home page, as well as the primary part of the manufacturer's Web site that discusses the device, must provide a prominently placed hyperlink to the Web page containing this information and must allow unrestricted viewing access. If the device can be purchased from the Web site or testing using the device can be ordered from the Web site, the same information must be found on the Web page for ordering the device or provided in a prominently placed and publicly accessible hyperlink on the Web page for ordering the device. Any changes to the device that could significantly affect safety or effectiveness would require new data or information in support of such changes, which would also have to be posted on the manufacturer's Web site. The information must include:
(i) A detailed device description including:
(A) Gene (or list of the genes if more than one) and variants the test detects (using standardized nomenclature, Human Genome Organization (HUGO) nomenclature, and coordinates).
(B) Scientifically established clinical validity of each variant detected and reported by the test, which must be well-established in peer-reviewed journal articles, authoritative summaries of the literature such as Genetics Home Reference (http://ghr.nlm.nih.gov/ ), GeneReviews (http://www.ncbi.nlm.nih.gov/books/NBK1116/ ), or similar summaries of valid scientific evidence, and/or professional society recommendations, including:
(1 ) Genotype-phenotype information for the reported mutations.
(2 ) Relevant American College of Medical Genetics (ACMG) or American Congress of Obstetricians and Gynecologists (ACOG) guideline recommending testing of the specific gene(s) and variants the test detects and recommended populations, if available. If not available, a statement stating that professional guidelines currently do not recommend testing for this specific gene(s) and variants.
(3 ) Table of expected prevalence of carrier status in major ethnic and racial populations and the general population.
(C) The specimen type (e.g., saliva, whole blood), matrix, and volume.
(D) Assay steps and technology used.
(E) Specification of required ancillary reagents, instrumentation, and equipment.
(F) Specification of the specimen collection, processing, storage, and preparation methods.
(G) Specification of risk mitigation elements and description of all additional procedures, methods, and practices incorporated into the directions for use that mitigate risks associated with testing.
(H) Information pertaining to the probability of test failure (e.g., failed quality control) based on data from clinical samples, description of scenarios in which a test can fail (i.e., low sample volume, low DNA concentration, etc.), how customers will be notified, and followup actions to be taken.
(I) Specification of the criteria for test result interpretation and reporting.
(ii) Information that demonstrates the performance characteristics of the device, including:
(A) Accuracy (method comparison) of study results for each claimed specimen type.
(1 ) Accuracy of the device shall be evaluated with fresh clinical specimens collected and processed in a manner consistent with the device's instructions for use. If this is impractical, fresh clinical samples may be substituted or supplemented with archived clinical samples. Archived samples shall have been collected previously in accordance with the device's instructions for use, stored appropriately, and randomly selected. In some instances, use of contrived samples or human cell line samples may also be appropriate; the contrived or human cell line samples shall mimic clinical specimens as much as is feasible and provide an unbiased evaluation of the device's accuracy.
(2 ) Accuracy must be evaluated as compared to bidirectional sequencing or other methods identified as appropriate by FDA. Performance criteria for both the comparator method and device must be predefined and appropriate to the test's intended use. Detailed appropriate study protocols must be provided.
(3 ) Information provided shall include the number and type of specimens, broken down by clinically relevant variants, that were compared to bidirectional sequencing or other methods identified as appropriate by FDA. The accuracy, defined as positive percent agreement (PPA) and negative percent agreement (NPA), must be measured; accuracy point estimates must be greater than 99 percent (both per reported variant and overall) and uncertainty of the point estimate must be presented using the 95 percent confidence interval. Clinical specimens must include both homozygous wild type and heterozygous genotypes. The number of clinical specimens for each variant reported that must be included in the accuracy study must be based on the variant prevalence. Common variants (greater than 0.1 percent allele frequency in ethnically relevant population) must have at least 20 unique heterozygous clinical specimens tested. Rare variants (less than or equal to 0.1 percent allele frequency in ethnically relevant population) shall have at least three unique mutant heterozygous specimens tested. Any no calls (i.e., absence of a result) or invalid calls (e.g., failed quality control) in the study must be included in accuracy study results and reported separately. Variants that have a point estimate for PPA or NPA of less than 99 percent (incorrect test results as compared to bidirectional sequencing or other methods identified as appropriate by FDA) must not be incorporated into test claims and reports. Accuracy measures generated from clinical specimens versus contrived samples or cell lines must be presented separately. Results must be summarized and presented in tabular format, by sample and by genotype. Point estimate of PPA should be calculated as the number of positive results divided by the number of specimens known to harbor variants (mutations) without "no calls" or invalid calls. The point estimate of NPA should be calculated as the number of negative results divided by the number of wild type specimens tested without "no calls" or invalid calls, for each variant that is being reported. Point estimates should be calculated along with 95 percent two-sided confidence intervals.
(4 ) Information shall be reported on the clinical positive predictive value (PPV) and negative predictive value (NPV) for carrier status (and where possible, for each variant) in each population. Specifically, to calculate PPV and NPV, estimate test coverage (TC) and the percent of persons with variant(s) included in the device among all carriers: PPV = (PPA * TC * Ï?)/(PPA * TC * Ï? + (1 â?? NPA) * (1 â?? Ï?)) and NPV = (NPA * (1 â?? Ï?))/(NPA *(1 â?? Ï?) + (1 â?? PPA*TC) * Ï?) where PPA and NPA described either in paragraph (b)(3)(ii)(A)(4 )(i ) or in paragraph (b)(3)(ii)(A)(4 )(ii ) of this section and Ï? is prevalence of carriers in the population (pre-test risk to be a carrier for the disease).
(i ) For the point estimates of PPA and NPA less than 100 percent, use the calculated estimates in the PPV and NPV calculations.
(ii ) Point estimates of 100 percent may have high uncertainty. If these variants are measured using highly multiplexed technology, calculate the random error rate for the overall device and incorporate that rate in the estimation of the PPA and NPA as calculated previously. Then use these calculated estimates in the PPV and NPV calculations. This type of accuracy study is helpful in determining that there is no systematic error in such devices.
(B) Precision (reproducibility): Precision data must be generated using multiple instruments and multiple operators, on multiple non-consecutive days, and using multiple reagent lots. The sample panel must include specimens with claimed sample type (e.g. saliva samples) representing different genotypes (i.e., wild type, heterozygous). Performance criteria must be predefined. A detailed study protocol must be created in advance of the study and then followed. The "failed quality control" rate must be indicated. It must be clearly documented whether results were generated from clinical specimens, contrived samples, or cell lines. The study results shall state, in a tabular format, the variants tested in the study and the number of replicates for each variant, and what testing conditions were studied (i.e., number of runs, days, instruments, reagent lots, operators, specimens/type, etc). The study must include all nucleic acid extraction steps from the claimed specimen type or matrix, unless a separate extraction study for the claimed sample type is performed. If the device is to be used at more than one laboratory, different laboratories must be included in the precision study (and reproducibility must be evaluated). The percentage of "no calls" or invalid calls, if any, in the study must be provided as a part of the precision (reproducibility) study results.
(C) Analytical specificity data: Data must be generated evaluating the effect on test performance of potential endogenous and exogenous interfering substances relevant to the specimen type, evaluation of cross-reactivity of known cross-reactive alleles and pseudogenes, and assessment of cross-contamination.
(D) Analytical sensitivity data: Data must be generated demonstrating the minimum amount of DNA that will enable the test to perform accurately in 95 percent of runs.
(E) Device stability data: The manufacturer must establish upper and lower limits of input nucleic acid and sample stability that will achieve the claimed accuracy and reproducibility. Data supporting such claims must be described.
(F) Specimen type and matrix comparison data: Specimen type and matrix comparison data must be generated if more than one specimen type or anticoagulant can be tested with the device, including failure rates for the different specimen types.
(iii) If the device is offered over-the-counter, including cases in which the test results are provided direct-to-consumer, the manufacturer must conduct a study that assesses user comprehension of the device's labeling and test process and provide a concise summary of the results of the study. The following items must be included in the user study:
(A) The test manufacturer must perform pre- and post-test user comprehension studies to assess user ability to understand the possible results of a carrier test and their clinical meaning. The comprehension test questions must directly evaluate the material being presented to the user in the test reports.
(B) The test manufacturer must provide a carrier testing education module to potential and actual test report recipients. The module must define terms that are used in the test reports and explain the significance of carrier status.
(C) The user study must meet the following criteria:
(1 ) The study participants must be comprised of a statistically justified and demographically diverse population (determined using methods such as quota-based sampling) that is representative of the intended user population. Furthermore, the users must be comprised of a diverse range of age and educational levels that have no prior experience with the test or its manufacturer. These factors shall be well-defined in the inclusion and exclusion criteria.
(2 ) All sources of bias (e.g., non-responders) must be predefined and accounted for in the study results with regard to both responders and non-responders.
(3 ) The testing must follow a format where users have limited time to complete the studies (such as an onsite survey format and a one-time visit with a cap on the maximum amount of time that a participant has to complete the tests).
(4 ) Users must be randomly assigned to study arms. Test reports given to users must: Define the condition being tested and related symptoms; explain the intended use and limitations of the test; explain the relevant ethnicities regarding the variant tested; explain carrier status and relevance to the user's ethnicity; and provide links to additional information pertaining to situations where the user is concerned about their test results or would like followup information as indicated in test labeling. The study shall assess participants' ability to understand the following comprehension concepts: The test's limitations, purpose, and results.
(5 ) Study participants must be untrained, naive to the test subject of the study, and be provided only the materials that will be available to them when the test is marketed.
(6 ) The user comprehension study must meet the predefined primary endpoint criteria, including a minimum of a 90 percent or greater overall comprehension rate (i.e. selection of the correct answer) for each comprehension concept to demonstrate that the education module and test reports are adequate for over-the-counter use.
(D) A summary of the user comprehension study must be provided and include the following:
(1 ) Results regarding reports that are provided for each gene/variant/ethnicity tested.
(2 ) Statistical methods used to analyze all data sets.
(3 ) Completion rate, non-responder rate, and reasons for non-response/data exclusion, as well as a summary table of comprehension rates regarding comprehension concepts (purpose of test, test results, test limitations, ethnicity relevance for the test results, etc.) for each study report.
(4) Your 21 CFR 809.10 compliant labeling and any test report generated must include the following warning and limitation statements, as applicable:
(i) A warning that reads "The test is intended only for autosomal recessive carrier screening in adults of reproductive age."
(ii) A statement accurately disclosing the genetic coverage of the test in lay terms, including, as applicable, information on variants not queried by the test, and the proportion of incident disease that is not related to the gene(s) tested. For example, where applicable, the statement would have to include a warning that the test does not or may not detect all genetic variants related to the genetic disease, and that the absence of a variant tested does not rule out the presence of other genetic variants that may be disease-related. Or, where applicable, the statement would have to include a warning that the basis for the disease for which the genetic carrier status is being tested is unknown or believed to be non-heritable in a substantial number of people who have the disease, and that a negative test result cannot rule out the possibility that any offspring may be affected with the disease. The statement would have to include any other warnings needed to accurately convey to consumers the degree to which the test is informative for carrier status.
(iii) For prescription use tests, the following warnings that read:
(A) "The results of this test are intended to be interpreted by a board-certified clinical molecular geneticist or equivalent and should be used in conjunction with other available laboratory and clinical information."
(B) "This device is not intended for disease diagnosis, prenatal testing of fetuses, risk assessment, prognosis or pre-symptomatic testing, susceptibility testing, or newborn screening."
(iv) For over-the-counter tests, a statement that reads "This test is not intended to diagnose a disease, or tell you anything about your risk for developing a disease in the future. On its own, this test is also not intended to tell you anything about the health of your fetus, or your newborn child's risk of developing a particular disease later on in life."
(v) For over-the-counter tests, the following warnings that read:
(A) "This test is not a substitute for visits to a healthcare provider. It is recommended that you consult with a healthcare provider if you have any questions or concerns about your results."
(B) "The test does not diagnose any health conditions. Results should be used along with other clinical information for any medical purposes."
(C) "The laboratory may not be able to process your sample. The probability that the laboratory cannot process your saliva sample can be up to [actual probability percentage]."
(D) "Your ethnicity may affect how your genetic health results are interpreted."
(vi) For a positive result in an over-the-counter test when the positive predictive value for a specific population is less than 50 percent and more than 5 percent, a warning that reads "The positive result you obtained may falsely identify you as a carrier. Consider genetic counseling and followup testing."
(vii) For a positive result in an over-the-counter test when the positive predictive value for a specific population is less than 5 percent, a warning that reads "The positive result you obtained is very likely to be incorrect due to the rarity of this variant. Consider genetic counseling and followup testing."
(5) The testing done to comply with paragraph (b)(3) of this section must show the device meets or exceeds each of the following performance specifications:
(i) The accuracy must be shown to be equal to or greater than 99 percent for both PPA and NPA. Variants that have a point estimate for PPA or NPA of less than 99 percent (incorrect test results as compared to bidirectional sequencing or other methods identified as appropriate by FDA) must not be incorporated into test claims and reports.
(ii) Precision (reproducibility) performance must meet or exceed 99 percent for both positive and negative results.
(iii) The user comprehension study must obtain values of 90 percent or greater user comprehension for each comprehension concept.
(6) The distribution of this device, excluding the collection device described in paragraph (b)(2) of this section, shall be limited to the manufacturer, the manufacturer's subsidiaries, and laboratories regulated under the Clinical Laboratory Improvement Amendments.
[80 FR 65630, Oct. 27, 2015, as amended at 82 FR 51570, Nov. 7, 2017]
|
|
|
Sec. 866.5950 Genetic health risk assessment system.
|
|
(a) Identification. A genetic health risk assessment system is a qualitative in vitro molecular diagnostic system used for detecting variants in genomic deoxyribonucleic acid (DNA) isolated from human specimens that will provide information to users about their genetic risk of developing a disease to inform lifestyle choices and/or conversations with a health care professional. This assessment system is for over-the-counter use. This device does not determine the person's overall risk of developing a disease.
(b) Classification. Class II (special controls). The genetic health risk assessment system device, when it has previously received a first-time FDA marketing authorization (e.g., 510(k) clearance) for the genetic health risk assessment system (a "one-time FDA reviewed genetic health risk assessment system"), is exempt from the premarket notification procedures in part 807, subpart E, of this chapter subject to the limitations in § 866.9. The device must comply with the following special controls:
(1) The 21 CFR 809.10 compliant labeling and any prepurchase page and test report generated, unless otherwise specified, must include:
(i) A section addressed to users with the following information:
(A) The limiting statement explaining that this test provides genetic risk information based on assessment of specific genetic variants but does not report on a user's entire genetic profile. This test [does not/may not, as appropriate] detect all genetic variants related to a given disease, and the absence of a variant tested does not rule out the presence of other genetic variants that may be related to the disease.
(B) The limiting statement explaining that other companies offering a genetic risk test may be detecting different genetic variants for the same disease, so the user may get different results using a test from a different company.
(C) The limiting statement explaining that other factors such as environmental and lifestyle risk factors may affect the risk of developing a given disease.
(D) The limiting statement explaining that some people may feel anxious about getting genetic test health results. This is normal. If the potential user feels very anxious, such user should speak to his or her doctor or other health care professional prior to collection of a sample for testing. This test is not a substitute for visits to a doctor or other health care professional. Users should consult with their doctor or other health care professional if they have any questions or concerns about the results of their test or their current state of health.
(E) Information about how to obtain access to a genetic counselor, board-certified clinical molecular geneticist, or equivalent health care professional about the results of a user's test.
(F) The limiting statement explaining that this test is not intended to diagnose a disease, tell you anything about your current state of health, or be used to make medical decisions, including whether or not you should take a medication or how much of a medication you should take.
(G) A limiting statement explaining that the laboratory may not be able to process a sample, and a description of the next steps to be taken by the manufacturer and/or the customer, as applicable.
(ii) A section in your 21 CFR 809.10 labeling and any test report generated that is for health care professionals who may receive the test results from their patients with the following information:
(A) The limiting statement explaining that this test is not intended to diagnose a disease, determine medical treatment, or tell the user anything about their current state of health.
(B) The limiting statement explaining that this test is intended to provide users with their genetic information to inform lifestyle decisions and conversations with their doctor or other health care professional.
(C) The limiting statement explaining that any diagnostic or treatment decisions should be based on testing and/or other information that you determine to be appropriate for your patient.
(2) The genetic test must use a sample collection device that is FDA-cleared, -approved, or -classified as 510(k) exempt, with an indication for in vitro diagnostic use in over-the-counter DNA testing.
(3) The device's labeling must include a hyperlink to the manufacturer's public Web site where the manufacturer shall make the information identified in paragraph (b)(3) of this section publicly available. The manufacturer's home page, as well as the primary part of the manufacturer's Web site that discusses the device, must provide a hyperlink to the Web page containing this information and must allow unrestricted viewing access. If the device can be purchased from the Web site or testing using the device can be ordered from the Web site, the same information must be found on the Web page for ordering the device or provided in a publicly accessible hyperlink on the Web page for ordering the device. Any changes to the device that could significantly affect safety or effectiveness would require new data or information in support of such changes, which would also have to be posted on the manufacturer's Web site. The information must include:
(i) An index of the material being provided to meet the requirements in paragraph (b)(3) of this section and its location.
(ii) A section that highlights summary information that allows the user to understand how the test works and how to interpret the results of the test. This section must, at a minimum, be written in plain language understandable to a lay user and include:
(A) Consistent explanations of the risk of disease associated with all variants included in the test. If there are different categories of risk, the manufacturer must provide literature references that support the different risk categories. If there will be multiple test reports and multiple variants, the risk categories must be defined similarly among them. For example, "increased risk" must be defined similarly between different test reports and different variant combinations.
(B) Clear context for the user to understand the context in which the cited clinical performance data support the risk reported. This includes, but is not limited to, any risks that are influenced by ethnicity, age, gender, environment, and lifestyle choices.
(C) Materials that explain the main concepts and terminology used in the test that include:
(1 ) Definitions: Scientific terms that are used in the test reports.
(2 ) Prepurchase page: This page must contain information that informs the user about what information the test will provide. This includes, but is not limited to, variant information, the condition or disease associated with the variant(s), professional guideline recommendations for general genetic risk testing, the limitations associated with the test (e.g., test does not detect all variants related to the disease) and any precautionary information about the test the user should be aware of before purchase. When the test reports the risk of a life-threatening or irreversibly debilitating disease or condition for which there are few or no options to prevent, treat, or cure the disease, a user opt-in section must be provided. This opt-in page must be provided for each disease that falls into this category and must provide specific information relevant to each test result. The opt-in page must include:
(i ) An option to accept or decline to receive this specific test result;
(ii ) Specification of the risk involved if the user is found to have the specific genetic test result;
(iii ) Professional guidelines that recommend when genetic testing for the associated target condition is or is not recommended; and
(iv ) A recommendation to speak with a health care professional, genetic counselor, or equivalent professional before getting the results of the test.
(3 ) Frequently asked questions (FAQ) page: This page must provide information that is specific for each variant/disease pair that is reported. Information provided in this section must be scientifically valid and supported by corresponding publications. The FAQ page must explain the health condition/disease being tested, the purpose of the test, the information the test will and will not provide, the relevance of race and ethnicity to the test results, information about the population to which the variants in the test is most applicable, the meaning of the result(s), other risk factors that contribute to disease, appropriate followup procedures, how the results of the test may affect the user's family, including children, and links to resources that provide additional information.
(iii) A technical information section containing the following information:
(A) Gene(s) and variant(s) the test detects using standardized nomenclature, Human Genome Organization nomenclature and coordinates as well as Single Nucleotide Polymorphism Database (dbSNP) reference SNP numbers (rs#).
(B) Scientifically established disease-risk association of each variant detected and reported by the test. This risk association information must include:
(1 ) Genotype-phenotype information for the reported variants.
(2 ) Table of expected frequency and risks of developing the disease in relevant ethnic populations and the general population.
(3 ) A statement about the current professional guidelines for testing these specific gene(s) and variant(s).
(i ) If professional guidelines are available, provide the recommendations in the professional guideline for the gene, variant, and disease, for when genetic testing should or should not be performed, and cautionary information that should be communicated when a particular gene and variant is detected.
(ii ) If professional guidelines are not available, provide a statement that the professional guidelines are not available for these specific gene(s) and variant(s).
(C) The specimen type (e.g., saliva, capillary whole blood).
(D) Assay steps and technology used.
(E) Specification of required ancillary reagents, instrumentation, and equipment.
(F) Specification of the specimen collection, processing, storage, and preparation methods.
(G) Specification of risk mitigation elements and description of all additional procedures, methods, and practices incorporated into the directions for use that mitigate risks associated with testing.
(H) Information pertaining to the probability of test failure (i.e., percentage of tests that failed quality control) based on data from clinical samples, a description of scenarios in which a test can fail (i.e., low sample volume, low DNA concentration, etc.), how users will be notified of a test failure, and the nature of followup actions on a failed test to be taken by the user and the manufacturer.
(I) Specification of the criteria for test result interpretation and reporting.
(J) Information that demonstrates the performance characteristics of the test, including:
(1 ) Accuracy of study results for each claimed specimen type.
(i ) Accuracy of the test shall be evaluated with fresh clinical specimens collected and processed in a manner consistent with the test's instructions for use. If this is impractical, fresh clinical samples may be substituted or supplemented with archived clinical samples. Archived samples shall have been collected previously in accordance with the instructions for use, stored appropriately, and randomly selected. In some limited circumstances, use of contrived samples or human cell line samples may also be appropriate and used as an acceptable alternative. The contrived or human cell line samples shall mimic clinical specimens as much as is feasible and provide an unbiased evaluation of the device accuracy.
(ii ) Accuracy must be evaluated by comparison to bidirectional Sanger sequencing or other methods identified as appropriate by FDA. Performance criteria for both the comparator method and the device must be predefined and appropriate to the device's intended use. Detailed study protocols must be provided.
(iii ) Test specimens must include all genotypes that will be included in the tests and reports. The number of samples tested in the accuracy study for each variant reported must be based on the variant frequency using either the minimum numbers of samples identified in this paragraph or, when determined appropriate and identified by FDA, a minimum number of samples determined using an alternative method. When appropriate, the same samples may be used in testing to demonstrate the accuracy of testing for multiple genotypes by generating sequence information at multiple relevant genetic locations. At least 20 unique samples representing the wild-type genotype must be tested. To test samples that are heterozygous for the reported variant(s), common variants (>0.1 percent variant frequency in the relevant population) must be tested with at least 20 unique samples. Rare variants (â?¤0.1 percent variant frequency in the relevant population) must be tested with at least three unique samples. To test samples that are homozygous for the reported variant(s), variants with â?¥2 percent variant frequency in a relevant population must be tested with at least 20 unique samples. Variants with a frequency in the relevant population <2 percent and â?¥0.5 percent must be tested with at least 10 unique samples. Variants with a frequency in the relevant population <0.5 percent must be tested with at least three unique samples. If variants with a frequency of <0.5 percent are not found within the relevant population and homozygous samples are not tested, then the test results for this homozygous rare variant must not be reported to the user.
(iv ) Information about the accuracy study shall include the number and type of samples that were compared to bidirectional Sanger sequencing or other methods identified as appropriate by FDA. This information must either be reported in tabular format and arranged by clinically relevant variants or reported using another method identified as appropriate by FDA. As an example, for samples with different genotypes DD, Dd, and dd, the following table represents data from the accuracy study presented in tabular format:

(v ) The accuracy represents the degrees of agreement between the device results and the comparator results. The accuracy must be evaluated by measuring different percent agreements (PA) of device results with the comparator results and percent of `no calls' or `invalid calls.' Calculate the rate of `no calls' and `invalid calls' for each comparator output as %Inv(DD) = A4/NDD, %Inv(Dd) = B4/NDd, %Inv(dd) = C4/Ndd. If `no calls' or `invalid calls' are required to be retested according to the device instructions for use, the percent of final `no calls' or `invalid calls' must be provided. In the table presenting the results of the accuracy study, use only the final results (i.e., after retesting the initial `no calls' or `invalid calls', if required according to the instructions for use). Samples that resulted in a `no call' or `invalid call' after retesting must not be included in the final calculations of agreement. If the percentages of `no calls' or `invalid calls' for each comparator output are similar, combine these estimates as (A4 + B4 + C4)/(NDD + NDd + Ndd) and provide a 95 percent two-sided confidence interval. The percent of final `no calls' or `invalid calls' must be clinically acceptable.
(vi ) Point estimates of percent agreement for each genotype must be calculated as the number of correct calls for that genotype divided by the number of samples known to contain that genotype excluding `no calls' or `invalid calls'. The calculations must be performed as follows:
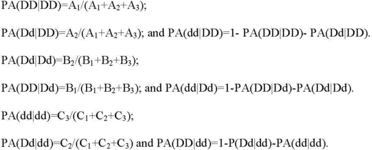
(vii ) For percent agreements for DD, Dd and dd (PA(DD|DD), PA(Dd|Dd) and PA(dd|dd)) as described in paragraph (b)(3)(iii)(J)(1 )(vi ) of this section, the 95 percent two-sided confidence intervals must be provided. The accuracy point estimates for percent agreements for DD, Dd and dd must be â?¥99 percent per reported variant and overall. Any variants that have a point estimate for either PA(DD|DD), PA(Dd|Dd), or PA(dd|dd) of <99 percent compared to bidirectional sequencing or other methods identified as appropriate by FDA must not be incorporated into test claims and reports. Accuracy results generated from clinical specimens versus contrived samples or cell lines must be presented separately. Results must be summarized and presented in tabular format by sample type and by genotype or must be reported using another method identified as appropriate by FDA (see paragraph (b)(3)(iii)(J)(1 )(iv ) of this section).
(viii ) Information must be reported on the Technical Positive Predictive Value (TPPV) related to the analytical (technical) performance of the device for genotypes in each relevant subpopulation (e.g., ethnicity, gender, age, geographical location, etc.). TPPV is the percentage of individuals with the genotype truly present among individuals whose test reports indicate that this genotype is present. The TPPV depends on the accuracy measures of percent agreements and on the frequency of the genotypes in the subpopulation being studied. The f(DD) is the frequency of DD and f(Dd) is the frequency of Dd in the subpopulation being studied; TPPV must be calculated as described in paragraphs (b)(3)(iii)(J)(1 )(ix ) through (xi ) of this section.
(ix ) For variants where the point estimates of PA(DD|DD), PA(Dd|Dd) and PA(dd|dd) are less than 100 percent, use these point estimates in TPPV calculations.
(x ) Point estimates of 100 percent in the accuracy study may have high uncertainty about performance of the test in the population. If these variants are measured using highly multiplexed technology, calculate the random error rate for the overall device. The accuracy study described in paragraph (b)(3)(iii)(J) of this section in those cases is more to determine that there is no systematic error in such devices. In those cases, incorporate that rate in the estimation of the percent agreements as calculated in paragraph (b)(3)(iii)(J)(1 )(vi ) of this section and include it in TPPV calculations.
(xi ) The TPPV for subpopulations with genotype frequencies of f(dd), f(Dd) and f(DD) = 1â??f(dd)â??f(Dd) in the subpopulation is calculated as:

(2 ) Precision and reproducibility data must be provided using multiple instruments and multiple operators, on multiple non-consecutive days, and using multiple reagent lots. The sample panel must either include specimens from the claimed sample type (e.g., saliva) representing all genotypes for each variant (e.g., wild type, heterozygous, and homozygous) or, if an alternative panel composition of specimens is identified by FDA as appropriate, a panel composed of those specimens FDA identified as appropriate. A detailed study protocol must be created in advance of the study and must include predetermined acceptance criteria for performance results. The percentage of samples that failed quality control must be indicated (i.e., the total number of sample replicates for which a sequence variant cannot be called (no calls) or that fail sequencing quality control criteria divided by the total number of replicates tested). It must be clearly documented whether results were generated from clinical specimens, contrived samples, or cell lines. The study results shall report the variants tested in the study and the number of replicates for each variant, and what conditions were tested (i.e., number of runs, days, instruments, reagent lots, operators, specimens/type, etc.). Results must be evaluated and presented in tabular format and stratified by study parameter (e.g., by site, instrument(s), reagent lot, operator, and sample variant). The study must include all extraction steps from the claimed specimen type or matrix, unless a separate extraction reproducibility study for the claimed sample type is performed. If the device is to be used at more than one laboratory, different laboratories must be included in the reproducibility study and reproducibility across sites must be evaluated. Any no calls or invalid calls in the study must be listed as a part of the precision and reproducibility study results.
(3 ) Analytical specificity data: Data must be provided that evaluates the effect of potential endogenous and exogenous interferents on test performance, including specimen extraction and variant detection. Interferents tested must include those reasonably likely to be potentially relevant to the sample type used for the device.
(4 ) Interfering variant data: Nucleotide mutations that can interfere with the technology must be cited and evaluated. Data must be provided to demonstrate the effect of the interfering variant(s) on the performance of the correct calls. Alternatively, for each suspected interfering mutation for which data is not provided demonstrating the effect of the interfering variant, the manufacturer must identify the suspected interfering variants in the labeling and indicate that the impact that the interfering variants may have on the assay's performance has not been studied by providing a statement that reads "It is possible that the presence of [insert clearly identifying information for the suspected interfering variant] in a sample may interfere with the performance of this test. However, its effect on the performance of this test has not been studied."
(5 ) Analytical sensitivity data: Data must be provided demonstrating the minimum amount of DNA that will enable the test to perform correctly in 95 percent of runs.
(6 ) Reagent stability: The manufacturer must evaluate reagent stability using wild-type, heterozygous, and homozygous samples. Reagent stability data must demonstrate that the reagents maintain the claimed accuracy and reproducibility. Data supporting such claims must be provided.
(7 ) Specimen type and matrix comparison data: Specimen type and matrix comparison data must be generated if more than one specimen type can be tested with this device, including failure rates for the different specimens.
(K) Clinical performance summary.
(1 ) Information to support the clinical performance of each variant reported by the test must be provided.
(2 ) Manufacturers must organize information by the specific variant combination as appropriate (e.g., wild type, heterozygous, homozygous, compound heterozygous, hemizygous genotypes). For each variant combination, information must be provided in the clinical performance section to support clinical performance for the risk category (e.g., not at risk, increased risk). For each variant combination, a summary of key results must be provided in tabular format or using another method identified as appropriate by FDA to include the appropriate information regarding variant type, data source, definition of the target condition (e.g., disease), clinical criteria for determining whether the target disease is present or absent, description of subjects with the target disease present and target disease absent (exclusion or inclusion criteria), and technical method for genotyping. When available, information on the effect of the variant on risk must be provided as the risk of a disease (lifetime risk or lifetime incidences) for an individual compared with the general population risk.
(i ) If odds ratios are available, using information about the genotype distribution either among individuals with the target disease absent, or in the general population, or information about the risk variant frequency and odds ratios, the likelihood ratios for the corresponding device results along with 95 percent confidence intervals must be calculated. Using information about pretest risk (Ï?), an estimate of likelihood ratio (LR), and a relationship between post-test risk R as R/(1â??R) = LR-Ï?/(1â??Ï?), the post-test risk R must be calculated.
(ii ) When available, likelihood ratios (LR) for different test results must be presented in a tabular format along with references to the source data or using another method identified as appropriate by FDA as stated in paragraph (b)(3)(iii)(K)(2) of this section. When these values are not directly available in published literature, likelihood ratios can be separately calculated along with the 95 percent confidence interval with references to the source data. Note that a minimum requirement for the presence of the variant's effect on the risk is that a corresponding LR is statistically higher than 1 (a lower bound of 95 percent two-sided confidence interval is larger than 1). It means that the post-test risk is statistically higher than the pretest risk (an observed value of the difference between the post-test and pretest risks).
(L) Materials that explain the main concepts and terminology used in the test that includes, but is not limited to:
(1 ) Definitions: Scientific terms that are used in the test reports.
(2 ) Prepurchase page: This page must contain information that informs the user about what the test will provide. This includes, but is not limited to, variant information, the condition or disease associated with the variant(s), professional guideline recommendations for general genetic risk testing, the limitations associated with the test (e.g., test does not detect all variants related to the disease) and any precautionary information about the test the user should be aware of before purchase. When the test reports the risk of a life-threatening or irreversibly debilitating disease or condition for which there are few or no options to prevent, treat, or cure the disease, a user opt-in section must be provided. This opt-in page must be provided for each disease that falls into this category and must provide specific information relevant to each test result. The opt-in page must include:
(i ) An option to accept or decline to receive this specific test result;
(ii ) Specification of the risk involved if the user is found to have the specific genetic test result;
(iii ) Professional guidelines that recommend when genetic testing for the associated target condition is or is not recommended; and
(iv ) A recommendation to speak with a health care professional, genetic counselor, or equivalent professional before getting the results of the test.
(3 ) Frequently asked questions (FAQ) page: This page must provide information that is specific for each variant/disease pair that is reported. Information provided in this section must be scientifically valid and supported by corresponding publications. The FAQ page must explain the health condition/disease being tested, the purpose of the test, the information the test will and will not provide, the relevance of race and ethnicity on the test results, information about the population to which the variants in the test is most applicable, the meaning of the result(s), other risks factors that contribute to disease, appropriate followup procedures, how the results of the test may affect the user's family, including children, and links to resources that provide additional information.
(M) User comprehension study: Information on a study that assesses comprehension of the test process and results by potential users of the test must be provided.
(1 ) The test manufacturer must provide a genetic risk education module to naive user comprehension study participants prior to their participation in the user comprehension study. The module must define terms that are used in the test reports and explain the significance of genetic risk reports.
(2 ) The test manufacturer must perform pre- and post-test user comprehension studies. The comprehension test questions must include directly evaluating a representative sample of the material being presented to the user as described in paragraph (b)(3)(ii) of this section.
(3 ) The manufacturer must provide a justification from a physician and/or genetic counselor that identifies the appropriate general and variant-specific concepts contained within the material being tested in the user comprehension study to ensure that all relevant concepts are incorporated in the study.
(4 ) The user study must meet the following criteria:
(i ) The study participants must comprise a statistically sufficient sample size and demographically diverse population (determined using methods such as quota-based sampling) that is representative of the intended user population. Furthermore, the study participants must comprise a diverse range of age and educational levels and have no prior experience with the test or its manufacturer. These factors shall be well defined in the inclusion and exclusion criteria.
(ii ) All sources of bias must be predefined and accounted for in the study results with regard to both responders and non-responders.
(iii ) The testing must follow a format where users have limited time to complete the studies (such as an onsite survey format and a one-time visit with a cap on the maximum amount of time that a participant has to complete the tests).
(iv ) Users must be randomly assigned to study arms. Test reports in the user comprehension study given to users must define the target condition being tested and related symptoms, explain the intended use and limitations of the test, explain the relevant ethnicities in regard to the variant tested, explain genetic health risks and relevance to the user's ethnicity, and assess participants' ability to understand the following comprehension concepts: The test's limitations, purpose, appropriate action, test results, and other factors that may have an impact on the test results.
(v ) Study participants must be untrained, be naive to the test subject of the study, and be provided the labeling prior to the start of the user comprehension study.
(vi ) The user comprehension study must meet the predefined primary endpoint criteria, including a minimum of a 90 percent or greater overall comprehension rate (i.e., selection of the correct answer) for each comprehension concept. Other acceptance criteria may be acceptable depending on the concept being tested. Meeting or exceeding this overall comprehension rate demonstrates that the materials presented to the user are adequate for over-the-counter use.
(vii ) The analysis of the user comprehension results must include results regarding reports that are provided for each gene/variant/ethnicity tested, statistical methods used to analyze all data sets, and completion rate, non-responder rate, and reasons for nonresponse/data exclusion. A summary table of comprehension rates regarding comprehension concepts (e.g., purpose of test, test results, test limitations, ethnicity relevance for the test results, etc.) for each study report must be included.
(4) The intended use of the device must not include the following indications for use:
(i) Prenatal testing;
(ii) Determining predisposition for cancer where the result of the test may lead to prophylactic screening, confirmatory procedures, or treatments that may incur morbidity or mortality to the patient;
(iii) Assessing the presence of genetic variants that impact the metabolism, exposure, response, risk of adverse events, dosing, or mechanisms of prescription or over-the-counter medications; or
(iv) Assessing the presence of deterministic autosomal dominant variants.
[82 FR 51561, Nov. 7, 2017, as amended at 83 FR 25914, June 5, 2018]
|
|
|
Sec. 866.5960 Human leukocyte antigen typing companion diagnostic test.
|
|
(a) Identification. A human leukocyte antigen (HLA) typing companion diagnostic (CDx) test is a prescription genotyping or phenotyping in vitro diagnostic product intended for use as an aid in identifying patients who have specific HLA allele(s) or express specific HLA antigen(s) and may benefit from treatment with a corresponding therapeutic product or are likely to be at increased risk for serious adverse reactions as a result of treatment with a corresponding therapeutic product.
(b) Classification. Class II (special controls). The special controls for this device are:
(1) The intended use of the device must specify the target HLA allele(s) or antigen(s), the patient population(s), and the corresponding therapeutic product(s).
(2) Design verification and validation must include:
(i) Detailed documentation of an analytical accuracy study that uses well-characterized samples including clinical samples from intended use population(s) focusing on the target allele(s) needed for patient selection;
(ii) Detailed documentation of precision studies (repeatability, reproducibility) that evaluate possible sources of variation that may affect test results;
(iii) Detailed documentation of a study determining range of input sample concentrations that meet performance specifications;
(iv) Detailed description of the ambiguity resolution method, if applicable;
(v) For a sequencing-based assay, documentation of coverage and predefined coverage threshold of target genomic regions, pertinent variant types, and sequence contexts;
(vi) For multiplex assays, documentation of a risk assessment and design specifications that are in place to prevent incorrect reactivity assignment;
(vii) Description of a plan on how to ensure the performance of the device does not change when new HLA alleles are identified, and/or when reactivity assignments are changed; and
(viii) Detailed description of device software including standalone software, or software and bioinformatics analysis pipeline, if applicable, incorporated in the instruments, and documentation of software including the level of concern and associated risks, software requirement specifications, software design specifications (e.g., algorithms, alarms and device limitations), hazard analysis, traceability matrix, verification and validation testing, unresolved anomalies, hardware requirements, and effective cybersecurity management.
(3) Clinical validity data (which may include summary reports from clinical trials, comparison studies using clinical samples, or through an alternative approach determined to be appropriate by FDA), demonstrating the following, as applicable:
(i) Which patients identified by the HLA CDx test are most likely to benefit from the corresponding therapeutic product; and
(ii) Which patients identified by the HLA CDx test are likely to be at increased risk for serious adverse reactions as a result of treatment with the corresponding therapeutic product.
(4) If the HLA test used in the clinical trials is different from the HLA CDx test in the premarket notification submission, the submission must include results of a bridging study, or an alternative approach determined to be appropriate by FDA.
[87 FR 79252, Dec. 27, 2022]
|
|
|
Authority: 21 U.S.C. 351, 360, 360c, 360e, 360j, 360l, 371.
Source: 47 FR 50823, Nov. 9, 1982, unless otherwise noted.
|
|





