|
Mineral oil may be safely used as a component of nonfood articles intended for use in contact with food, subject to the provisions of this section:
(a) White mineral oil meeting the specifications prescribed in § 172.878 of this chapter may be used as a component of nonfood articles provided such use complies with any applicable limitations in parts 170 through 189 of this chapter. The use of white mineral oil in or on food itself, including the use of white mineral oil as a protective coating or release agent for food, is subject to the provisions of § 172.878 of this chapter.
(b) Technical white mineral oil identified in paragraph (b)(1) of this section may be used as provided in paragraph (b)(2) of this section.
(1) Technical white mineral oil consists of specially refined distillates of virgin petroleum or of specially refined distillates that are produced synthetically from petroleum gases. Technical white mineral oil meets the following specifications:
(i) Saybolt color 20 minimum as determined by ASTM method D156-82, "Standard Test Method for Saybolt Color of Petroleum Products (Saybolt Chromometer Method)," which is incorporated by reference. Copies may be obtained from the American Society for Testing Materials, 100 Barr Harbor Dr., West Conshohocken, Philadelphia, PA 19428-2959, or may be examined at the National Archives and Records Administration (NARA). For information on the availability of this material at NARA, call 202-741-6030, or go to: http://www.archives.gov/federal_register/code_of_federal_regulations/ibr_locations.html.
(ii) Ultraviolet absorbance limits as follows:
| Wavelength (m[micro])
| Maximum absorbance per centimeter optical pathlength
|
|---|
| 280 to 289 | 4.0
| | 290 to 299 | 3.3
| | 300 to 329 | 2.3
| | 330 to 350 | 0.8 |
Technical white mineral oil containing antioxidants shall meet the specified ultraviolet absorbance limits after correction for any absorbance due to the antioxidants. The ultraviolet absorbance shall be determined by the procedure described for application to mineral oil under "Specification" on page 66 of the "Journal of the Association of Official Agricultural Chemists," Volume 45 (February 1962) (which is incorporated by reference; copies are available from the Center for Food Safety and Applied Nutrition (HFS-200), Food and Drug Administration, 5001 Campus Dr., College Park, MD 20740, or available for inspection at the National Archives and Records Administration (NARA). For information on the availability of this material at NARA, call 202-741-6030, or go to: http://www.archives.gov/federal_register/code_of_federal_regulations/ibr_locations.html. ), disregarding the last two sentences of that procedure and substituting therefor the following: Determine the absorbance of the mineral oil extract in a 10-millimeter cell in the range from 260-350 m[micro], inclusive, compared to the solvent control. If the absorbance so measured exceeds 2.0 at any point in range 280-350 m[micro], inclusive, dilute the extract and the solvent control, respectively, to twice their volume with dimethyl sulfoxide and remeasure the absorbance. Multiply the remeasured absorbance values by 2 to determine the absorbance of the mineral oil extract per centimeter optical pathlength.
(2) Technical white mineral oil may be used wherever mineral oil is permitted for use as a component of nonfood articles complying with §§ 175.105, 176.200, 176.210, 177.2260, 177.2600, and 177.2800 of this chapter and §§ 178.3570 and 178.3910.
(3) Technical white mineral oil may contain any antioxidant permitted in food by regulations issued in accordance with section 409 of the Act, in an amount not greater than that required to produce its intended effect.
(c) Mineral oil identified in paragraph (c)(1) of this section may be used as provided in paragraph (c)(2) of this section.
(1) The mineral oil consists of virgin petroleum distillates refined to meet the following specifications:
(i) Initial boiling point of 450 deg.F minimum.
(ii) Color 5.5 maximum as determined by ASTM method D1500-82, "Standard Test Method for ASTM Color of Petroleum Products (ASTM Color Scale)," which is incorporated by reference. The availability of this incorporation by reference is given in paragraph (b)(1)(i) of this section.
(iii) Ultraviolet absorbance limits as follows as determined by the analytical method described in paragraph (c)(3) of this section:
| Wavelength (m[micro])
| Maximum absorbance per centimeter optical pathlength
|
|---|
| 280 to 289 | 0.7
| | 290 to 299 | 0.6
| | 300 to 359 | 0.4
| | 360 to 400 | .09 |
(2) The mineral oil may be used wherever mineral oil is permitted for use as a component of nonfood articles complying with §§ 175.105 and 176.210 of this chapter and § 178.3910 (for use only in rolling of metallic foil and sheet stock), §§ 176.200, 177.2260, 177.2600, and 177.2800 of this chapter.
(3) The analytical method for determining ultraviolet absorbance limit is as follows:
general instructions
Because of the sensitivity of the test, the possibility of errors arising from contamination is great. It is of the greatest importance that all glassware be scrupulously cleaned to remove all organic matter such as oil, grease, detergent residues, etc. Examine all glassware, including stoppers and stopcocks, under ultraviolet light to detect any residual fluorescent contamination. As a precautionary measure it is recommended practice to rinse all glassware with purified isooctane immediately before use. No grease is to be used on stopcocks or joints. Great care to avoid contamination of oil samples in handling and to assure absence of any extraneous material arising from inadequate packaging is essential. Because some of the polynuclear hydrocarbons sought in this test are very susceptible to photo-oxidation, the entire procedure is to be carried out under subdued light.
apparatus
Separatory funnels. 250-milliliter, 500-milliliter, 1,000-milliliter, and preferably 2,000-milliliter capacity, equipped with tetrafluoroethylene polymer stopcocks.
Reservoir. 500-milliliter capacity, equipped with a 24/40 standard taper male fitting at the bottom and a suitable ball-joint at the top for connecting to the nitrogen supply. The male fitting should be equipped with glass hooks.
Chromatographic tube. 180 millimeters in length, inside diameter to be 15.7 millimeters +/-0.1 millimeter, equipped with a coarse, fritted-glass disc, a tetrafluoroethylene polymer stopcock, and a female 24/40 standard tapered fitting at the opposite end. (Overall length of the column with the female joint is 235 millimeters.) The female fitting should be equipped with glass hooks.
Disc. Tetrafluoroethylene polymer 2-inch diameter disk approximately
3/16-inch thick with a hole bored in the center to closely fit the stem of the chromatographic tube.
Suction flask. 250-milliliter or 500-milliliter filter flask.
Condenser. 24/40 joints, fitted with a drying tube, length optional.
Evaporation flask (optional). 250-milliliter or 500-milliliter capacity all-glass flask equipped with standard taper stopper having inlet and outlet tubes to permit passage of nitrogen across the surface of contained liquid to be evaporated.
Spectrophotometric cells. Fused quartz cells, optical path length in the range of 5,000 centimeter +/-0.005 centimeter; also for checking spectrophotometer performance only, optical path length in the range 1,000 centimeter +/-0.005 centimeter. With distilled water in the cells, determine any absorbance differences.
Spectrophotometer. Spectral range 250 millimicrons - 400 millimicrons with spectral slit width of 2 millimicrons or less; under instrument operating conditions for these absorbance measurements, the spectrophotometer shall also meet the following performance requirements:
Absorbance repeatability, +/-0.01 at 0.4 absorbance.
Absorbance accuracy
1
+/-0.05 at 0.4 absorbance.
Wavelength accuracy, +/-1.0 millimicron.
Nitrogen cylinder. Water-pumped or equivalent purity nitrogen in cylinder equipped with regulator and valve to control flow at 5 p.s.i.g.
reagents and materials
Organic solvents. All solvents used throughout the procedure shall meet the specifications and tests described in this specification. The isooctane, benzene, acetone, and methyl alcohol designated in the list following this paragraph shall pass the following test:
To the specified quantity of solvent in a 250-milliliter Erlenmeyer flask, add 1 milliliter of purified n- hexadecane and evaporate on the steam bath under a stream of nitrogen (a loose aluminum foil jacket around the flask will speed evaporation). Discontinue evaporation when not over 1 milliliter of residue remains. (To the residue from benzene add a 10-milliliter portion of purified isooctane, reevaporate, and repeat once to insure complete removal of benzene.)
Alternatively, the evaporation time can be reduced by using the optional evaporation flask. In this case the solvent and n- hexadecane are placed in the flask on the steam bath, the tube assembly is inserted, and a stream of nitrogen is fed through the inlet tube while the outlet tube is connected to a solvent trap and vacuum line in such a way as to prevent any flow-back of condensate into the flask.
Dissolve the 1 milliliter of hexadecane residue in isooctane and make to 25 milliliters volume. Determine the absorbance in the 5-centimeter path length cells compared to isooctane as reference. The absorbance of the solution of the solvent residue (except for methyl alcohol) shall not exceed 0.01 per centimeter path length between 280 and 400 m[micro]. For methyl alcohol this absorbance value shall be 0.00.
Isooctane (2,2,4-trimethylpentane). Use 180 milliliters for the test described in the preceding paragraph. Purify, if necessary, by passage through a column of activated silica gel (Grade 12, Davison Chemical Company, Baltimore, Maryland, or equivalent) about 90 centimeters in length and 5 centimeters to 8 centimeters in diameter.
Benzene, A.C.S. reagent grade. Use 150 milliliters for the test. Purify, if necessary, by distillation or otherwise.
Acetone, A.C.S. reagent grade. Use 200 milliliters for the test. Purify, if necessary, by distillation.
Eluting mixtures:
1. 10 percent benzene in isooctane. Pipet 50 milliliters of benzene into a 250-milliliter glass-stoppered volumetric flask and adjust to volume with isooctane, with mixing.
2. 20 percent benzene in isooctane. Pipet 50 milliliters of benzene into a 250-milliliter glass-stoppered volumetric flask and adjust to volume with isooctane, with mixing.
3. Acetone-benzene-water mixture. Add 20 milliliters of water to 380 milliliters of acetone and 200 milliliters of benzene, and mix.
n-Hexadecane, 99-percent olefin-free. Dilute 1.0 milliliter of n- hexadecane to 25 milliliters with isooctane and determine the absorbance in a 5-centimeter cell compared to isooctane as reference point between 280 m[micro]-400 m[micro]. The absorbance per centimeter path length shall not exceed 0.00 in this range. Purify, if necessary, by percolation through activated silica gel or by distillation.
Methyl alcohol, A.C.S. reagent grade. Use 10.0 milliliters of methyl alcohol. Purify, if necessary, by distillation.
Dimethyl sulfoxide. Spectrophotometric grade (Crown Zellerbach Corporation, Camas, Washington, or equivalent). Absorbance (1-centimeter cell, distilled water reference, sample completely saturated with nitrogen).
| Wavelength
| Absorbance (maximum)
|
|---|
| 261.5 | 1.00
| | 270 | .20
| | 275 | .09
| | 280 | .06
| | 300 | .015 |
There shall be no irregularities in the absorbance curve within these wavelengths.
Phosphoric acid. 85 percent A.C.S. reagent grade.
Sodium borohydride. 98 percent.
Magnesium oxide (Sea Sorb 43, Food Machinery Company, Westvaco Division, distributed by chemical supply firms, or equivalent). Place 100 grams of the magnesium oxide in a large beaker, add 700 milliliters of distilled water to make a thin slurry, and heat on a steam bath for 30 minutes with intermittent stirring. Stir well initially to insure that all the adsorbent is completely wetted. Using a Buchner funnel and a filter paper (Schleicher & Schuell No. 597, or equivalent) of suitable diameter, filter with suction. Continue suction until water no longer drips from the funnel. Transfer the adsorbent to a glass trough lined with aluminum foil (free from rolling oil). Break up the magnesia with a clean spatula and spread out the adsorbent on the aluminum foil in a layer about 1 centimeter to 2 centimeters thick. Dry for 24 hours at 160 deg.C +/-1 deg.C. Pulverize the magnesia with mortar and pestle. Sieve the pulverized adsorbent between 60-180 mesh. Use the magnesia retained on the 180-mesh sieve.
Celite 545. Johns Mansville Company, diatomaceous earth, or equivalent.
Magnesium oxide-Celite 545 mixture (2 + 1) by weight. Place the magnesium oxide (60-180 mesh) and the Celite 545 in 2 to 1 proportions, respectively, by weight in a glass-stoppered flask large enough for adequate mixing. Shake vigorously for 10 minutes. Transfer the mixture to a glass trough lined with aluminum foil (free from rolling oil) and spread it out on a layer about 1 centimeter to 2 centimeters thick. Reheat the mixture at 160 deg.C +/-1 deg.C for 2 hours, and store in a tightly closed flask.
Sodium sulfate, anhydrous, A.C.S. reagent grade, preferably in granular form. For each bottle of sodium sulfate reagent used, establish as follows the necessary sodium sulfate prewash to provide such filters required in the method: Place approximately 35 grams of anhydrous sodium sulfate in a 30-milliliter course, fritted-glass funnel or in a 65-millimeter filter funnel with glass wool plug; wash with successive 15-milliliter portions of the indicated solvent until a 15-milliliter portion of the wash shows 0.00 absorbance per centimeter path length between 280 m[micro] and 400 m[micro] when tested as prescribed under "Organic solvents." Usually three portions of wash solvent are sufficient.
Before proceeding with analysis of a sample, determine the absorbance in a 5-centimeter path cell between 250 millimicrons and 400 millimicrons for the reagent blank by carrying out the procedure, without an oil sample, recording the spectra after the extraction stage and after the complete procedure as prescribed. The absorbance per centimeter pathlength following the extraction stage should not exceed 0.02 in the wavelength range from 280 m[micro] to 400 m[micro]; the absorbance per centimeter pathlength following the complete procedure should not exceed 0.02 in the wavelength range from 280 m[micro] to 400 m[micro]. If in either spectrum the characteristic benzene peaks in the 250 m[micro]-260 m[micro] region are present, remove the benzene by the procedure under "Organic solvents" and record absorbance again.
Place 300 milliliters of dimethyl sulfoxide in a 1-liter separatory funnel and add 75 milliliters of phosphoric acid. Mix the contents of the funnel and allow to stand for 10 minutes. (The reaction between the sulfoxide and the acid is exothermic. Release pressure after mixing, then keep funnel stoppered.) Add 150 milliliters of isooctane and shake to pre-equilibrate the solvents. Draw off the individual layers and store in glass-stoppered flasks.
Weigh a 20-gram sample of the oil and transfer to a 500-milliliter separatory funnel containing 100 milliliters of pre-equilibrated sulfoxide-phosphoric acid mixture. Complete the transfer of the sample with small portions of preequilibrated isooctane to give a total volume of the oil and solvent of 75 milliliters. Shake the funnel vigorously for 2 minutes. Set up three 250-milliliter separatory funnels with each containing 30 milliliters of pre-equilibrated isooctane. After separation of liquid phases, carefully draw off lower layer into the first 250-milliliter separatory funnel and wash in tandem with the 30-milliliter portions of isooctane contained in the 250-milliliter separatory funnels. Shaking time for each wash is 1 minute. Repeat the extraction operation with two additional portions of the sulfoxide-acid mixture and wash each extractive in tandem through the same three portions of isooctane.
Collect the successive extractives (300 milliliters total) in a separatory funnel (preferably 2-liter) containing 480 milliliters of distilled water; mix, and allow to cool for a few minutes after the last extractive has been added. Add 80 milliliters of isooctane to the solution and extract by shaking the funnel vigorously for 2 minutes. Draw off the lower aqueous layer into a second separatory funnel (preferably 2-liter) and repeat the extraction with 80 milliliters of isooctane. Draw off and discard the aqueous layer. Wash each of the 80-milliliter extractives three times with 100-milliliter portions of distilled water. Shaking time for each wash is 1 minute. Discard the aqueous layers. Filter the first extractive through anhydrous sodium sulfate prewashed with isooctane (see Sodium sulfate under "Reagents and Materials" for preparation of filter) into a 250-milliliter Erlenmeyer flask (or optionally into the evaporation flask). Wash the first separatory funnel with the second 80-milliliter isooctane extractive and pass through the sodium sulfate. Then wash the second and first separatory funnels successively with a 20-milliliter portion of isooctane and pass the solvent through the sodium sulfate into the flask. Add 1 milliliter of n -hexadecane and evaporate the isooctane on the steam bath under nitrogen. Discontinue evaporation when not over 1 milliliter of residue remains. To the residue, add a 10-milliliter portion of isooctane, reevaporate to 1 milliliter of hexadecane, and repeat this operation once.
Quantitatively transfer the residue with isooctane to a 200-milliliter volumetric flask, make to volume, and mix. Determine the absorbance of the solution in the 1-centimeter pathlength cells compared to isooctane as reference between 280 m[micro]-400 m[micro] (take care to lose none of the solution in filling the sample cell). Correct the absorbance values for any absorbance derived from reagents as determined by carrying out the procedure without an oil sample. If the corrected absorbance does not exceed the limits prescribed in this paragraph, the oil meets the ultraviolet absorbance specifications. If the corrected absorbance per centimeter pathlength exceeds the limits prescribed in this paragraph, proceed as follows: Quantitatively transfer the isooctane solution to a 125-milliliter flask equipped with 24/40 joint, and evaporate the isooctane on the steam bath under a stream of nitrogen to a volume of 1 milliliter of hexadecane. Add 10 milliliters of methyl alcohol and approximately 0.3 gram of sodium borohydride. (Minimize exposure of the borohydride to the atmosphere. A measuring dipper may be used.) Immediately fit a water-cooled condenser equipped with a 24/40 joint and with a drying tube into the flask, mix until the borohydride is dissolved, and allow to stand for 30 minutes at room temperature, with intermittent swirling. At the end of this period, disconnect the flask and evaporate the methyl alcohol on the steam bath under nitrogen until the sodium borohydride begins to come out of the solution. Then add 10 milliliters of isooctane and evaporate to a volume of about 2-3 milliliters. Again, add 10 milliliters of isooctane and concentrate to a volume of approximately 5 milliliters. Swirl the flask repeatedly to assure adequate washing of the sodium borohydride residues.
Fit the tetrafluoroethylene polymer disc on the upper part of the stem of the chromatographic tube, then place the tube with the disc on the suction flask and apply the vacuum (approximately 135 millimeters Hg pressure). Weigh out 14 grams of the 2:1 magnesium oxide-Celite 545 mixture and pour the adsorbent mixture into the chromatographic tube in approximately 3-centimeter layers. After the addition of each layer, level off the top of the adsorbent with a flat glass rod or metal plunger by pressing down firmly until the adsorbent is well packed. Loosen the topmost few millimeters of each adsorbent layer with the end of a metal rod before the addition of the next layer. Continue packing in this manner until all the 14 grams of the adsorbent is added to the tube. Level off the top of the adsorbent by pressing down firmly with a flat glass rod or metal plunger to make the depth of the adsorbent bed approximately 12.5 centimeters in depth. Turn off the vacuum and remove the suction flask. Fit the 500-milliliter reservoir onto the top of the chromatographic column and prewet the column by passing 100 milliliters of isooctane through the column. Adjust the nitrogen pressure so that the rate of descent of the isooctane coming off the column is between 2-3 milliliters per minute. Discontinue pressure just before the last of the isooctane reaches the level of the adsorbent. (Caution: Do not allow the liquid level to recede below the adsorbent level at any time.) Remove the reservoir and decant the 5-milliliter isooctane concentrate solution onto the column and with slight pressure again allow the liquid level to recede to barely above the adsorbent level. Rapidly complete the transfer similarly with two 5-milliliter portions of isooctane, swirling the flask repeatedly each time to assure adequate washing of the residue. Just before the final 5-milliliter wash reaches the top of the adsorbent, add 100 milliliters of isooctane to the reservoir and continue the percolation at the 2-3 milliliters per minute rate. Just before the last of the isooctane reaches the adsorbent level, add 100 milliliters of 10 percent benzene in isooctane to the reservoir and continue the percolation at the aforementioned rate. Just before the solvent mixture reaches adsorbent level, add 25 milliliters of 20 percent benzene in isooctane to the reservoir and continue the percolation at 2-3 milliliters per minute until all this solvent mixture has been removed from the column. Discard all the elution solvents collected up to this point. Add 300 milliliters of the acetone-benzene-water mixture to the reservoir and percolate through the column to eluate the polynuclear compounds. Collect the eluate in a clean 1-liter separatory funnel. Allow the column to drain until most of the solvent mixture is removed. Wash the eluate three times with 300-milliliter portions of distilled water, shaking well for each wash. (The addition of small amounts of sodium chloride facilitates separation.) Discard the aqueous layer after each wash. After the final separation, filter the residual benzene through anhydrous sodium sulfate pre-washed with benzene (see Sodium sulfate under "Reagents and Materials" for preparation of filter) into a 250-milliliter Erlenmeyer flask (or optionally into the evaporation flask). Wash the separatory funnel with two additional 20-milliliter portions of benzene which are also filtered through the sodium sulfate. Add 1 milliliter of n -hexadecane and completely remove the benzene by evaporation under nitrogen, using the special procedure to eliminate benzene as previously described under "Organic solvents." Quantitatively transfer the residue with isooctane to a 200-milliliter volumetric flask and adjust to volume. Determine the absorbance of the solution in the 1-centimeter pathlength cells compared to isooctane as reference between 250 m[micro]-400 m[micro]. Correct for any absorbance derived from the reagents as determined by carrying out the procedure without an oil sample. If either spectrum shows the characteristic benzene peaks in the 250 m[micro]-260 m[micro] region, evaporate the solution to remove benzene by the procedure under "Organic solvents." Dissolve the residue, transfer quantitatively, and adjust to volume in isooctane in a 200-milliliter volumetric flask. Record the absorbance again. If the corrected absorbance does not exceed the limits proposed in this paragraph, the oil meets the proposed ultraviolet absorbance specifications.
(d) Mineral oil identified in paragraph (d)(1) of this section may be used as provided in paragraph (d)(2) of this section.
(1) The mineral oil consists of virgin petroleum distillates refined to meet the following specifications:
(i) Distillation endpoint at 760 millimeters pressure not to exceed 371 deg.C, with a maximum residue not to exceed 2 percent, as determined by ASTM method D86-82, "Standard Method for Distillation of Petroleum Products," which is incorporated by reference. The availability of this incorporation by reference is given in paragraph (b)(1)(i) of this section.
(ii) Ultraviolet absorbance limits as follows as determined by the method described in paragraph (d)(3) of this section.
| Wavelength (m[micro])
| Maximum absorb-ance per centimeter optical pathlength
|
|---|
| 280 to 299 | 2.3
| | 300 to 319 | 1.2
| | 320 to 359 | .8
| | 360 to 400 | .3 |
(iii) Pyrene content not to exceed a maximum of 25 parts per million as determined by the method described in paragraph (d)(3) of this section.
(2) The mineral oil may be used only in the processing of jute fiber employed in the production of textile bags intended for use in contact with the following types of food: Dry grains and dry seeds (for example, beans, peas, rice, and lentils); whole root crop vegetables of the types identified in 40 CFR 180.34(f); unshelled and shelled nuts (including peanuts); and dry animal feed. The finished processed jute fiber shall contain no more than 6 percent by weight of residual mineral oil.
(3) The analytical method for determining ultraviolet absorbance limits and pyrene content is as follows:
I. Apparatus. A. Assorted beakers, separatory funnels fitted with tetrafluoroethylene polymer stopcocks, and graduated cylinders.
B. Volumetric flasks, 200-milliliter.
C. A chromatographic column made from nominal 1.3 centimeters outside diameter * 75 centimeters glass tubing tapered at one end and joined to a 2-millimeter-bore tetrafluoroethylene polymer stopcock. The opposite end is flanged and joined to a female 24/40 standard taper fitting. This provides for accommodating the 500-milliliter reservoir described in item I.E below.
D. A chromatographic column made from nominal 1.7 centimeters outside diameter * 115 centimeters glass tubing tapered at one end and joined to a 2-millimeter-bore tetrafluoroethylene polymer stopcock. The opposite end is flanged and joined to a 2.5 centimeters outside diameter * 9.0 centimeters glass tube having a female 24/40 standard taper fitting. This provides for accommodating the 500-milliliter reservoir described in item I. E below.
E. A 500-milliliter reservoir having a 24/40 standard taper male fitting at bottom and a suitable ball joint at the top for connecting to the nitrogen supply. The female fitting of the chromatographic columns described in items I. C and D above and the male fitting of the reservoir described in this item E should both be equipped with glass hooks.
(Note: Rubber stoppers are not to be used. Stopcock grease is not to be used on ground-glass joints in this method.)
F. A spectrophotometer equipped to automatically record absorbance of liquid samples in 1-centimeter pathlength cells in the spectral region of 280-400 m[micro] with a spectral slit width of 2 m[micro] or less. At an absorbance level of about 0.4, absorbance measurements shall be repeatable within +/-0.01 and accurate within +/-0.05. Wavelength measurements shall be repeatable with +/-0.2 m[micro] and accurate within +/-1.0 m[micro]. Instrument operating conditions are selected to realize this performance under dynamic (automatic) recording operations. Accuracy of absorbance measurements are determined at 290, 345, and 400 m[micro], using potassium chromate as the reference standard. (National Bureau of Standards Circular 484, Spectrophotometry, U.S. Department of Commerce, 1949.)
G. Two fused quartz cells having pathlengths of 1.00+/-0.005 centimeter or better.
II. Purity of reagents and materials. Reagent-grade chemicals shall be used in all tests. It is further specified that each chemical shall be tested for purity in accordance with the instruction given under "Reagents and Materials" in III below. In addition, a blank run by the procedure shall be made on each purified lot of reagents and materials. Unless otherwise indicated, references to water shall be understood to mean distilled water.
III. Reagents and materials - A. Organic solvents. All solvents used throughout the procedure shall meet the specifications and tests described in this section III. The isooctane, benzene, cyclohexane, nitromethane, and n -hexadecane designated shall pass the following test: To the specified quantity of solvent in a 150-milliliter beaker, add 1 milliliter of purified n -hexadecane and evaporate on the steam bath under a stream of nitrogen. Discontinue evaporation when not over 1 milliliter of residue remains (to the residue from benzene and nitromethane add a 10-milliliter portion of purified isooctane, re-evaporate, and repeat once to insure complete removal of solvent). Dissolve the 1 milliliter of n -hexadecane residue in isooctane and make to 10-milliliter volume. Determine the absorbance in 1.0-centimeter pathlength cells compared to water as reference. The absorbance of the solution of solvent residue shall not exceed 0.05 between 280 and 400 m[micro].
1. Isooctane (2,2,4-trimethylpentane ). Use 240 milliliters for the above test. Purify, if necessary, by passage through a column of activated silica gel.
2. Benzene. Use 200 milliliters for the above test. Purify, if necessary, by distillation or otherwise.
3. Cyclohexane. Use 70 milliliters for the above test. Purify, if necessary, by distillation, silica gel percolation, or otherwise.
4. Nitromethane. Use 125 milliliters for the above test. Purify, if necessary, by distillation or otherwise.
5. n-Hexadecane. Determine the absorbance on this solvent directly. Purify, if necessary, by silica gel percolation or otherwise.
B. Other materials - 1. Pyrene standard reference. Pyrene, reagent grade, melting point range 150-152 deg.C. (Organic Chemical 3627, Eastman Kodak Co., Rochester, N.Y., or equivalent). The standard reference absorbance is the absorbance at 334 millimicrons of a standard reference solution of pyrene containing a concentration of 1.0 milligram per liter in purified isooctane measured against isooctane of the same spectral purity in 1.0-centimeter cells. (This absorbance will be approximately 0.28.)
2. Chrysene solution. Prepare a solution at a concentration of 5.0 milligrams per liter by dissolving 5.0 milligrams of chrysene in purified isooctane in a 1-liter volumetric flask. Adjust to volume with isooctane.
3. Nitrogen gas. Water pumped or equivalent purity, cylinder with regulator, and valve control flow at 5 p.s.i.
4. Silica gel. 100-200 mesh (Davison Chemical, Baltimore, Md., Grade 923, or equivalent), purified and activated by the following procedure: Place about 1 kilogram of silica gel in a large column and wash with contaminant-free benzene until a 200-milliliter sample of the benzene coming off the column will pass the ultraviolet absorption test for benzene. This test is performed as stipulated under "Organic solvents" in A under III above. When the silica gel has been sufficiently cleaned, activate the gel before use by placing the 1-kilogram batch in a shallow container in a layer no greater than 1 inch in depth and heating in an oven (Caution! Explosion Hazard) at 130 deg.C. for 16 hours, and store in a vacuum desiccator. Reheating about once a week is necessary if the silica gel is repeatedly removed from the desiccator.
5. Aluminum oxide (Aluminum Co. of America, Grade F-20, or equivalent grade ). 80-200 mesh, purified and activated by the following procedure: Place about 1 kilogram of aluminum oxide in a large column and wash with contaminant-free benzene until a 200-milliliter sample of the benzene coming off the column will pass the ultraviolet absorption test for benzene. This test is performed as stipulated under "Organic solvents" in A under III above. (Caution! Remove Benzene From Adsorbent Under Vacuum To Minimize Explosion Hazard in Subsequent Heating!) When the aluminum oxide has been sufficiently cleaned and freed of solvent, activate it before use by placing the 1-kilogram batch in a shallow container in a layer no greater than 1 inch in depth. Heat in an oven at 130 deg.C for 16 hours. Upon removal from heat, store at atmospheric pressure over 80 percent (by weight) sulfuric acid in a desiccator for at least 36 hours before use. This gives aluminum oxide with between 6 to 9.5 percent volatiles. This is determined by heating a weighed sample of the prepared aluminum oxide at 2,000 deg.F for 2 hours and then quickly reweighing. To insure the proper adsorptive properties of the aluminum oxide, perform the following test:
a. Weigh 50 grams +/-1 gram of the activated aluminum oxide and pack into the chromatographic column (1.3 centimeters * 75 centimeters) described under "Apparatus" in C under I above. Use glass wool at the column exit to prevent the aluminum oxide from passing through the column.
b. Place a 250-milliliter graduated cylinder under the column to measure the amount of eluate coming from the column.
c. Prewet the aluminum oxide by passing 40 milliliters of isooctane through the column. Adjust the nitrogen pressure so that the rate of descent of the isooctane coming off the column is between 1.5 to 2.5 milliliters per minute.
d. Just prior to the last of the isooctane reaching the top of the aluminum oxide bed, add 10 milliliters of the isooctane solution containing 5.0 milligrams of chrysene per liter.
e. Continue percolation until the isooctane is just above the aluminum oxide. Then add 200 milliliters of a mixture of benzene and isooctane (33
1/3 percent benzene and 66
2/3 percent isooctane by volume) to the reservoir and continue percolation.
f. Continue percolation, collecting the eluates (40 milliliters of the prewet solution, 10 milliliters of the sample solution, and 200 milliliters of the gradient solution) in the 250-milliliter graduated cylinder until the level of the gradient solution is just above the aluminum oxide. Add 200 milliliters of the eluting solution of benzene and isooctane (90 percent benzene and 10 percent isooctane by volume) to the column and continue collecting until a total of 250 milliliters of solution has been obtained. This may be discarded. Now begin to collect the final eluate.
g. Place a 100-milliliter graduated cylinder under the column and continue the percolation until a 100-milliliter eluate has been obtained.
h. Measure the amount of chrysene in this 100-milliliter fraction by ultraviolet analysis. If the aluminum oxide is satisfactory, more than 80 percent of the original amount of chrysene should be found in this fraction. (Note: If the amount of chrysene recovered is less than 80 percent, the original batch of aluminum oxide should be sieved between 100-160 mesh. Activation and testing of this sieved batch should indicate a satisfactory aluminum oxide for use.)
IV. Sampling. Precautions must be taken to insure that an uncontaminated sample of the mineral oil is obtained since ultraviolet absorption is very sensitive to small amounts of extraneous material contaminating the sample through careless handling.
V. Procedure. A. Blank. Before proceeding with the analysis of a sample, determine the absorbance of the solvent residues by carrying out the procedure without a sample.
B. Sample. 1. Weigh out 20.0 grams +/-0.1 gram of the mineral oil into a beaker and transfer to a 250-milliliter separatory funnel fitted with a tetrafluoroethylene polymer stopcock, using enough cyclohexane (25 milliliters) to give a final total volume of 50 milliliters (mineral oil plus cyclohexane).
2. Add 25 milliliters of nitromethane saturated with cyclohexane and shake by hand vigorously for 3 minutes. Recover the lower nitromethane layer in a 150-milliliter beaker containing 1 milliliter of n -hexadecane and evaporate on the steam bath under nitrogen. Repeat the extraction four more times, recovering each extract in the 150-milliliter beaker. Exercise care not to fill the beaker to such a capacity that solvent losses may occur. Evaporate the combined nitromethane extracts to 1 milliliter of n -hexadecane residue containing the nitromethane-soluble mineral oil extractives. (Note: Complete removal of the nitromethane is essential. This can be assured by two successive additions of 5 milliliters of isooctane and reevaporation.)
3. Remove the beaker from the steam bath and allow to cool.
4. Weigh 50 grams +/-1 gram of activated aluminum oxide and pack into the chromatographic column (1.3 centimeters * 75 centimeters) described under "Apparatus" in C under I above. (Note: A small plug of glass wool is placed at the column exit to prevent the aluminum oxide from passing through the column. After adding aluminum oxide, tap the column lightly to remove air voids. All percolations using aluminum oxide are performed under nitrogen pressure. The 500-milliliter reservoir described under "Apparatus" in E under I above is to be used to hold the elution solvents.)
5. Prewet the column by adding 40 milliliters of isooctane to the column. Adjust nitrogen pressure so that rate of descent of the isooctane coming off the column is 2.0 to 3.0 milliliters per minute. Be careful to maintain the level of solvent in the reservoir to prevent air from entering the aluminum oxide bed. New or additional solvent is added just before the last portion of the previous solvent enters the bed. To minimize possible photo-oxidation effects, the following procedures (steps 6 through 18) shall be carried out in subdued light.
6. Before the last of the isooctane reaches the top of the aluminum oxide bed, release the nitrogen pressure and turn off the stopcock on the column. Transfer the n -hexadecane residue from the 150-milliliter beaker from procedure step 3 above onto the column, using several washes of isooctane (total volume of washes should be no greater than 10-15 milliliters).
7. Open the stopcock and continue percolation until the isooctane is about 1 centimeter above the top of the aluminum oxide bed. Add 200 milliliters of isooctane to the reservoir, and continue the percolation at the specified rate.
8. Just before the isooctane surface reaches the top of the aluminum oxide bed, add 200 milliliters of a mixture of benzene and isooctane (33
1/3 percent benzene and 66
2/3 percent isooctane by volume) to the reservoir, and continue the percolation.
9. Just before the surface of this mixture reaches the top of the aluminum oxide bed, release the nitrogen pressure, turn off the stopcock, and discard all the elution solvents collected up to this point.
10. Add to the reservoir 300 milliliters of a mixture of benzene and isooctane (90 percent benzene and 10 percent isooctane by volume), place a 25-milliliter graduated cylinder under the column, continue the percolation until 20 milliliters of eluate has been collected, and then discard the eluate.
11. At this point, place a clean 250-milliliter Erlenmeyer flask under the column. Continue the percolation and collect all the remaining eluate.
(Note: Allow the column to drain completely. An increase in the nitrogen pressure may be necessary as the last of the solvent comes off the column.)
12. Place 1 milliliter of n -hexadecane into a 150-milliliter beaker. Place this onto a steam bath under a nitrogen stream and transfer in small portions the eluate from step 11 above. Wash out the Erlenmeyer flask with small amounts of benzene and transfer to the evaporation beaker. Evaporate until only 1 milliliter of hexadecane residue remains. (Note: Complete removal of the benzene is essential. This can be assured by two successive additions of 5 milliliters of isooctane and reevaporation.)
13. Remove the beaker from the steam bath and cool.
14. Place a sample of 113.5 grams activated 100- 200-mesh silica gel in a 500-milliliter glass-stoppered Erlenmeyer flask. Add to the silica gel 46.2 grams (41 milliliters) of nitromethane. Stopper and shake the flask vigorously until no lumps of silica gel are observed and then shake occasionally during a period of 1 hour. The resultant nitromethane-treated silica gel is 29 weight-percent nitro-methane and 71 weight-percent silica gel.
15. Place a small plug of glass wool in the tapered end of the 1.7 centimeters outside diameter * 115 centimeters column, described under "Apparatus" in D of I above, adjacent to the stopcock to prevent silica gel from passing through the stopcock. Pack the nitromethane-treated silica gel into the column, tapping lightly. The resultant silica gel bed should be about 95 centimeters in depth. Place into a flask 170 milliliters of isooctane saturated with nitromethane.
16. Place a 100-milliliter graduated cylinder under the column and transfer the residue from the beaker in procedure step 13 above with several washes of the 170 milliliters of isooctane, saturated with nitromethane, onto the top of the column. (Total volume of washes should be no greater than 10 to 15 milliliters.) Permit isooctane solution to enter the silica gel bed until the liquid level is at the top bed level. Place the remaining amount of the 170 milliliters of isooctane, saturated with nitromethane, in the reservoir above the bed for percolation through the silica gel. Apply nitrogen pressure to the top of the column, adjusting the pressure so that the isooctane is collected at the rate of 2.5 to 3.5 milliliters per minute, and percolate isooctane through the bed until a quantity of 75.0 milliliters of eluate is collected. Discard the 75 milliliters of eluate. Turn off the stopcock and add 250 milliliters of benzene to the reservoir above the bed. Use a 400-milliliter beaker to collect the remaining eluate.
17. Open the stopcock, renew the pressure, and percolate the remaining isooctane and benzene through the column eluting the remaining aromatics. Transfer the eluate in small portions from the 400 milliliter beaker to a 150-milliliter beaker containing 1 milliliter of n -hexadecane and evaporate on the steam bath under nitrogen. Rinse the 400-milliliter beaker well with small portions of isooctane to obtain a complete transfer.
(Note: Complete removal of the nitromethane and benzene is essential. This can be assured by successive additions of 5 milliliters of isooctane and reevaporation.)
18. Transfer the residue with several washes of isooctane into a 200-milliliter volumetric flask. Add isooctane to mark.
19. Record the spectrum of the sample solution in a 1-centimeter cell compared to isooctane from 270 to 400 m[micro]. After making necessary corrections in the spectrum for cell differences and for the blank absorbance, record the maximum absorbance in each of the wavelength intervals (m[micro]), 280-299, 300-319, 320-359, 360-400.
a. If the spectrum then shows no discernible peak corresponding to the absorbance maximum of the pyrene reference standard solution at 334 m[micro], the maximum absorbances in the respective wavelength intervals recorded shall not exceed those prescribed in paragraph (d)(1)(ii) of this section.
b. If such a peak is evident in the spectrum of the sample solution, and the spectrum as a whole is not incompatible with that of a pyrene contaminant yielding such a peak of the observed absorbance, calculate the concentration of pyrene that would yield this peak (334 m) by the base-line technique described in ASTM method E169-63 (Reapproved 1981), "Standard Recommended Practices for General Techniques of Ultraviolet Quantitative Analysis," which is incorporated by reference. The availability of this incorporation by reference is given in paragraph (b)(1)(i) of this section. Correct each of the maximum absorbances in the respective specified wavelength intervals by subtracting the absorbance due to pyrene, determined as follows:
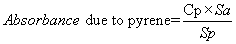
where:
Cp = Calculated concentration of pyrene in sample solution;
Sp = Concentration of pyrene reference standard solution in same units of concentration;
Sa = Absorbance of pyrene reference standard solution at wavelength of maximum absorbance of sample solution in the respective specified wavelength intervals.
Also calculate the pyrene content of the oil sample in parts per million as follows:
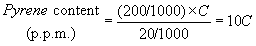
where:
C = Calculated concentration of pyrene in milligrams per liter of sample solution.
c. The pyrene content so determined shall not exceed 25 p.p.m. The maximum absorbances corrected for pyrene content as described in this step 19 for each of the specified wavelength intervals shall not exceed the limits prescribed in paragraph (d)(1)(ii) of this section.
d. If the spectrum as a whole of the sample solution is in any respect clearly incompatible with the presence of pyrene as the source of the peak at 334 m[micro], then the maximum absorbances in the respective wavelength intervals without correction for any assumed pyrene content shall not exceed the limits prescribed in paragraph (d)(1)(ii) of this section.
1 As determined by procedure using potassium chromate for reference standard and described in National Bureau of Standards Circular 484, Spectrophotometry, U.S. Department of Commerce (1949). The accuracy is to be determined by comparison with the standard values at 290, 345, and 400 millimicrons. Circular 484 is incorporated by reference. Copies are available from the Center for Food Safety and Applied Nutrition (HFS-200), Food and Drug Administration, 5001 Campus Dr., College Park, MD 20740, or available for inspection at the National Archives and Records Administration (NARA). For information on the availability of this material at NARA, call 202-741-6030, or go to: http://www.archives.gov/federal_register/code_of_federal_regulations/ibr_locations.html. [42 FR 14609, Mar. 15, 1977, as amended at 47 FR 11847, Mar. 19, 1982; 49 FR 10112, Mar. 19, 1984; 54 FR 24898, June 12, 1989]
| 




