|
(a) Identification. A genetic health risk assessment system is a qualitative in vitro molecular diagnostic system used for detecting variants in genomic deoxyribonucleic acid (DNA) isolated from human specimens that will provide information to users about their genetic risk of developing a disease to inform lifestyle choices and/or conversations with a health care professional. This assessment system is for over-the-counter use. This device does not determine the person's overall risk of developing a disease.
(b) Classification. Class II (special controls). The genetic health risk assessment system device, when it has previously received a first-time FDA marketing authorization (e.g., 510(k) clearance) for the genetic health risk assessment system (a "one-time FDA reviewed genetic health risk assessment system"), is exempt from the premarket notification procedures in part 807, subpart E, of this chapter subject to the limitations in § 866.9. The device must comply with the following special controls:
(1) The 21 CFR 809.10 compliant labeling and any prepurchase page and test report generated, unless otherwise specified, must include:
(i) A section addressed to users with the following information:
(A) The limiting statement explaining that this test provides genetic risk information based on assessment of specific genetic variants but does not report on a user's entire genetic profile. This test [does not/may not, as appropriate] detect all genetic variants related to a given disease, and the absence of a variant tested does not rule out the presence of other genetic variants that may be related to the disease.
(B) The limiting statement explaining that other companies offering a genetic risk test may be detecting different genetic variants for the same disease, so the user may get different results using a test from a different company.
(C) The limiting statement explaining that other factors such as environmental and lifestyle risk factors may affect the risk of developing a given disease.
(D) The limiting statement explaining that some people may feel anxious about getting genetic test health results. This is normal. If the potential user feels very anxious, such user should speak to his or her doctor or other health care professional prior to collection of a sample for testing. This test is not a substitute for visits to a doctor or other health care professional. Users should consult with their doctor or other health care professional if they have any questions or concerns about the results of their test or their current state of health.
(E) Information about how to obtain access to a genetic counselor, board-certified clinical molecular geneticist, or equivalent health care professional about the results of a user's test.
(F) The limiting statement explaining that this test is not intended to diagnose a disease, tell you anything about your current state of health, or be used to make medical decisions, including whether or not you should take a medication or how much of a medication you should take.
(G) A limiting statement explaining that the laboratory may not be able to process a sample, and a description of the next steps to be taken by the manufacturer and/or the customer, as applicable.
(ii) A section in your 21 CFR 809.10 labeling and any test report generated that is for health care professionals who may receive the test results from their patients with the following information:
(A) The limiting statement explaining that this test is not intended to diagnose a disease, determine medical treatment, or tell the user anything about their current state of health.
(B) The limiting statement explaining that this test is intended to provide users with their genetic information to inform lifestyle decisions and conversations with their doctor or other health care professional.
(C) The limiting statement explaining that any diagnostic or treatment decisions should be based on testing and/or other information that you determine to be appropriate for your patient.
(2) The genetic test must use a sample collection device that is FDA-cleared, -approved, or -classified as 510(k) exempt, with an indication for in vitro diagnostic use in over-the-counter DNA testing.
(3) The device's labeling must include a hyperlink to the manufacturer's public Web site where the manufacturer shall make the information identified in paragraph (b)(3) of this section publicly available. The manufacturer's home page, as well as the primary part of the manufacturer's Web site that discusses the device, must provide a hyperlink to the Web page containing this information and must allow unrestricted viewing access. If the device can be purchased from the Web site or testing using the device can be ordered from the Web site, the same information must be found on the Web page for ordering the device or provided in a publicly accessible hyperlink on the Web page for ordering the device. Any changes to the device that could significantly affect safety or effectiveness would require new data or information in support of such changes, which would also have to be posted on the manufacturer's Web site. The information must include:
(i) An index of the material being provided to meet the requirements in paragraph (b)(3) of this section and its location.
(ii) A section that highlights summary information that allows the user to understand how the test works and how to interpret the results of the test. This section must, at a minimum, be written in plain language understandable to a lay user and include:
(A) Consistent explanations of the risk of disease associated with all variants included in the test. If there are different categories of risk, the manufacturer must provide literature references that support the different risk categories. If there will be multiple test reports and multiple variants, the risk categories must be defined similarly among them. For example, "increased risk" must be defined similarly between different test reports and different variant combinations.
(B) Clear context for the user to understand the context in which the cited clinical performance data support the risk reported. This includes, but is not limited to, any risks that are influenced by ethnicity, age, gender, environment, and lifestyle choices.
(C) Materials that explain the main concepts and terminology used in the test that include:
(1 ) Definitions: Scientific terms that are used in the test reports.
(2 ) Prepurchase page: This page must contain information that informs the user about what information the test will provide. This includes, but is not limited to, variant information, the condition or disease associated with the variant(s), professional guideline recommendations for general genetic risk testing, the limitations associated with the test (e.g., test does not detect all variants related to the disease) and any precautionary information about the test the user should be aware of before purchase. When the test reports the risk of a life-threatening or irreversibly debilitating disease or condition for which there are few or no options to prevent, treat, or cure the disease, a user opt-in section must be provided. This opt-in page must be provided for each disease that falls into this category and must provide specific information relevant to each test result. The opt-in page must include:
(i ) An option to accept or decline to receive this specific test result;
(ii ) Specification of the risk involved if the user is found to have the specific genetic test result;
(iii ) Professional guidelines that recommend when genetic testing for the associated target condition is or is not recommended; and
(iv ) A recommendation to speak with a health care professional, genetic counselor, or equivalent professional before getting the results of the test.
(3 ) Frequently asked questions (FAQ) page: This page must provide information that is specific for each variant/disease pair that is reported. Information provided in this section must be scientifically valid and supported by corresponding publications. The FAQ page must explain the health condition/disease being tested, the purpose of the test, the information the test will and will not provide, the relevance of race and ethnicity to the test results, information about the population to which the variants in the test is most applicable, the meaning of the result(s), other risk factors that contribute to disease, appropriate followup procedures, how the results of the test may affect the user's family, including children, and links to resources that provide additional information.
(iii) A technical information section containing the following information:
(A) Gene(s) and variant(s) the test detects using standardized nomenclature, Human Genome Organization nomenclature and coordinates as well as Single Nucleotide Polymorphism Database (dbSNP) reference SNP numbers (rs#).
(B) Scientifically established disease-risk association of each variant detected and reported by the test. This risk association information must include:
(1 ) Genotype-phenotype information for the reported variants.
(2 ) Table of expected frequency and risks of developing the disease in relevant ethnic populations and the general population.
(3 ) A statement about the current professional guidelines for testing these specific gene(s) and variant(s).
(i ) If professional guidelines are available, provide the recommendations in the professional guideline for the gene, variant, and disease, for when genetic testing should or should not be performed, and cautionary information that should be communicated when a particular gene and variant is detected.
(ii ) If professional guidelines are not available, provide a statement that the professional guidelines are not available for these specific gene(s) and variant(s).
(C) The specimen type (e.g., saliva, capillary whole blood).
(D) Assay steps and technology used.
(E) Specification of required ancillary reagents, instrumentation, and equipment.
(F) Specification of the specimen collection, processing, storage, and preparation methods.
(G) Specification of risk mitigation elements and description of all additional procedures, methods, and practices incorporated into the directions for use that mitigate risks associated with testing.
(H) Information pertaining to the probability of test failure (i.e., percentage of tests that failed quality control) based on data from clinical samples, a description of scenarios in which a test can fail (i.e., low sample volume, low DNA concentration, etc.), how users will be notified of a test failure, and the nature of followup actions on a failed test to be taken by the user and the manufacturer.
(I) Specification of the criteria for test result interpretation and reporting.
(J) Information that demonstrates the performance characteristics of the test, including:
(1 ) Accuracy of study results for each claimed specimen type.
(i ) Accuracy of the test shall be evaluated with fresh clinical specimens collected and processed in a manner consistent with the test's instructions for use. If this is impractical, fresh clinical samples may be substituted or supplemented with archived clinical samples. Archived samples shall have been collected previously in accordance with the instructions for use, stored appropriately, and randomly selected. In some limited circumstances, use of contrived samples or human cell line samples may also be appropriate and used as an acceptable alternative. The contrived or human cell line samples shall mimic clinical specimens as much as is feasible and provide an unbiased evaluation of the device accuracy.
(ii ) Accuracy must be evaluated by comparison to bidirectional Sanger sequencing or other methods identified as appropriate by FDA. Performance criteria for both the comparator method and the device must be predefined and appropriate to the device's intended use. Detailed study protocols must be provided.
(iii ) Test specimens must include all genotypes that will be included in the tests and reports. The number of samples tested in the accuracy study for each variant reported must be based on the variant frequency using either the minimum numbers of samples identified in this paragraph or, when determined appropriate and identified by FDA, a minimum number of samples determined using an alternative method. When appropriate, the same samples may be used in testing to demonstrate the accuracy of testing for multiple genotypes by generating sequence information at multiple relevant genetic locations. At least 20 unique samples representing the wild-type genotype must be tested. To test samples that are heterozygous for the reported variant(s), common variants (>0.1 percent variant frequency in the relevant population) must be tested with at least 20 unique samples. Rare variants ( less than or equal to 0.1 percent variant frequency in the relevant population) must be tested with at least three unique samples. To test samples that are homozygous for the reported variant(s), variants with greater than or equal to 2 percent variant frequency in a relevant population must be tested with at least 20 unique samples. Variants with a frequency in the relevant population <2 percent and greater than or equal to 0.5 percent must be tested with at least 10 unique samples. Variants with a frequency in the relevant population <0.5 percent must be tested with at least three unique samples. If variants with a frequency of <0.5 percent are not found within the relevant population and homozygous samples are not tested, then the test results for this homozygous rare variant must not be reported to the user.
(iv ) Information about the accuracy study shall include the number and type of samples that were compared to bidirectional Sanger sequencing or other methods identified as appropriate by FDA. This information must either be reported in tabular format and arranged by clinically relevant variants or reported using another method identified as appropriate by FDA. As an example, for samples with different genotypes DD, Dd, and dd, the following table represents data from the accuracy study presented in tabular format:

(v ) The accuracy represents the degrees of agreement between the device results and the comparator results. The accuracy must be evaluated by measuring different percent agreements (PA) of device results with the comparator results and percent of `no calls' or `invalid calls.' Calculate the rate of `no calls' and `invalid calls' for each comparator output as %Inv(DD) = A4/NDD, %Inv(Dd) = B4/NDd, %Inv(dd) = C4/Ndd. If `no calls' or `invalid calls' are required to be retested according to the device instructions for use, the percent of final `no calls' or `invalid calls' must be provided. In the table presenting the results of the accuracy study, use only the final results (i.e., after retesting the initial `no calls' or `invalid calls', if required according to the instructions for use). Samples that resulted in a `no call' or `invalid call' after retesting must not be included in the final calculations of agreement. If the percentages of `no calls' or `invalid calls' for each comparator output are similar, combine these estimates as (A4 + B4 + C4)/(NDD + NDd + Ndd) and provide a 95 percent two-sided confidence interval. The percent of final `no calls' or `invalid calls' must be clinically acceptable.
(vi ) Point estimates of percent agreement for each genotype must be calculated as the number of correct calls for that genotype divided by the number of samples known to contain that genotype excluding `no calls' or `invalid calls'. The calculations must be performed as follows:
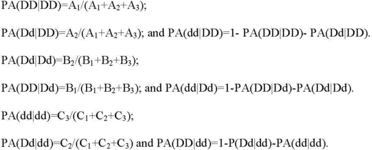
(vii ) For percent agreements for DD, Dd and dd (PA(DD|DD), PA(Dd|Dd) and PA(dd|dd)) as described in paragraph (b)(3)(iii)(J)(1 )(vi ) of this section, the 95 percent two-sided confidence intervals must be provided. The accuracy point estimates for percent agreements for DD, Dd and dd must be greater than or equal to 99 percent per reported variant and overall. Any variants that have a point estimate for either PA(DD|DD), PA(Dd|Dd), or PA(dd|dd) of <99 percent compared to bidirectional sequencing or other methods identified as appropriate by FDA must not be incorporated into test claims and reports. Accuracy results generated from clinical specimens versus contrived samples or cell lines must be presented separately. Results must be summarized and presented in tabular format by sample type and by genotype or must be reported using another method identified as appropriate by FDA (see paragraph (b)(3)(iii)(J)(1 )(iv ) of this section).
(viii ) Information must be reported on the Technical Positive Predictive Value (TPPV) related to the analytical (technical) performance of the device for genotypes in each relevant subpopulation (e.g., ethnicity, gender, age, geographical location, etc.). TPPV is the percentage of individuals with the genotype truly present among individuals whose test reports indicate that this genotype is present. The TPPV depends on the accuracy measures of percent agreements and on the frequency of the genotypes in the subpopulation being studied. The f(DD) is the frequency of DD and f(Dd) is the frequency of Dd in the subpopulation being studied; TPPV must be calculated as described in paragraphs (b)(3)(iii)(J)(1 )(ix ) through (xi ) of this section.
(ix ) For variants where the point estimates of PA(DD|DD), PA(Dd|Dd) and PA(dd|dd) are less than 100 percent, use these point estimates in TPPV calculations.
(x ) Point estimates of 100 percent in the accuracy study may have high uncertainty about performance of the test in the population. If these variants are measured using highly multiplexed technology, calculate the random error rate for the overall device. The accuracy study described in paragraph (b)(3)(iii)(J) of this section in those cases is more to determine that there is no systematic error in such devices. In those cases, incorporate that rate in the estimation of the percent agreements as calculated in paragraph (b)(3)(iii)(J)(1 )(vi ) of this section and include it in TPPV calculations.
(xi ) The TPPV for subpopulations with genotype frequencies of f(dd), f(Dd) and f(DD) = f(DD) = 1-f(dd)-f(Dd) in the subpopulation is calculated as:

(2 ) Precision and reproducibility data must be provided using multiple instruments and multiple operators, on multiple non-consecutive days, and using multiple reagent lots. The sample panel must either include specimens from the claimed sample type (e.g., saliva) representing all genotypes for each variant (e.g., wild type, heterozygous, and homozygous) or, if an alternative panel composition of specimens is identified by FDA as appropriate, a panel composed of those specimens FDA identified as appropriate. A detailed study protocol must be created in advance of the study and must include predetermined acceptance criteria for performance results. The percentage of samples that failed quality control must be indicated (i.e., the total number of sample replicates for which a sequence variant cannot be called (no calls) or that fail sequencing quality control criteria divided by the total number of replicates tested). It must be clearly documented whether results were generated from clinical specimens, contrived samples, or cell lines. The study results shall report the variants tested in the study and the number of replicates for each variant, and what conditions were tested (i.e., number of runs, days, instruments, reagent lots, operators, specimens/type, etc.). Results must be evaluated and presented in tabular format and stratified by study parameter (e.g., by site, instrument(s), reagent lot, operator, and sample variant). The study must include all extraction steps from the claimed specimen type or matrix, unless a separate extraction reproducibility study for the claimed sample type is performed. If the device is to be used at more than one laboratory, different laboratories must be included in the reproducibility study and reproducibility across sites must be evaluated. Any no calls or invalid calls in the study must be listed as a part of the precision and reproducibility study results.
(3 ) Analytical specificity data: Data must be provided that evaluates the effect of potential endogenous and exogenous interferents on test performance, including specimen extraction and variant detection. Interferents tested must include those reasonably likely to be potentially relevant to the sample type used for the device.
(4 ) Interfering variant data: Nucleotide mutations that can interfere with the technology must be cited and evaluated. Data must be provided to demonstrate the effect of the interfering variant(s) on the performance of the correct calls. Alternatively, for each suspected interfering mutation for which data is not provided demonstrating the effect of the interfering variant, the manufacturer must identify the suspected interfering variants in the labeling and indicate that the impact that the interfering variants may have on the assay's performance has not been studied by providing a statement that reads "It is possible that the presence of [insert clearly identifying information for the suspected interfering variant] in a sample may interfere with the performance of this test. However, its effect on the performance of this test has not been studied."
(5 ) Analytical sensitivity data: Data must be provided demonstrating the minimum amount of DNA that will enable the test to perform correctly in 95 percent of runs.
(6 ) Reagent stability: The manufacturer must evaluate reagent stability using wild-type, heterozygous, and homozygous samples. Reagent stability data must demonstrate that the reagents maintain the claimed accuracy and reproducibility. Data supporting such claims must be provided.
(7 ) Specimen type and matrix comparison data: Specimen type and matrix comparison data must be generated if more than one specimen type can be tested with this device, including failure rates for the different specimens.
(K) Clinical performance summary.
(1 ) Information to support the clinical performance of each variant reported by the test must be provided.
(2 ) Manufacturers must organize information by the specific variant combination as appropriate (e.g., wild type, heterozygous, homozygous, compound heterozygous, hemizygous genotypes). For each variant combination, information must be provided in the clinical performance section to support clinical performance for the risk category (e.g., not at risk, increased risk). For each variant combination, a summary of key results must be provided in tabular format or using another method identified as appropriate by FDA to include the appropriate information regarding variant type, data source, definition of the target condition (e.g., disease), clinical criteria for determining whether the target disease is present or absent, description of subjects with the target disease present and target disease absent (exclusion or inclusion criteria), and technical method for genotyping. When available, information on the effect of the variant on risk must be provided as the risk of a disease (lifetime risk or lifetime incidences) for an individual compared with the general population risk.
(i ) If odds ratios are available, using information about the genotype distribution either among individuals with the target disease absent, or in the general population, or information about the risk variant frequency and odds ratios, the likelihood ratios for the corresponding device results along with 95 percent confidence intervals must be calculated. Using information about pretest risk ([PI]), an estimate of likelihood ratio (LR), and a relationship between post-test risk R as R/(1â??R) = LR-[PI]/(1â??[PI]), the post-test risk R must be calculated.
(ii ) When available, likelihood ratios (LR) for different test results must be presented in a tabular format along with references to the source data or using another method identified as appropriate by FDA as stated in paragraph (b)(3)(iii)(K)(2) of this section. When these values are not directly available in published literature, likelihood ratios can be separately calculated along with the 95 percent confidence interval with references to the source data. Note that a minimum requirement for the presence of the variant's effect on the risk is that a corresponding LR is statistically higher than 1 (a lower bound of 95 percent two-sided confidence interval is larger than 1). It means that the post-test risk is statistically higher than the pretest risk (an observed value of the difference between the post-test and pretest risks).
(L) Materials that explain the main concepts and terminology used in the test that includes, but is not limited to:
(1 ) Definitions: Scientific terms that are used in the test reports.
(2 ) Prepurchase page: This page must contain information that informs the user about what the test will provide. This includes, but is not limited to, variant information, the condition or disease associated with the variant(s), professional guideline recommendations for general genetic risk testing, the limitations associated with the test (e.g., test does not detect all variants related to the disease) and any precautionary information about the test the user should be aware of before purchase. When the test reports the risk of a life-threatening or irreversibly debilitating disease or condition for which there are few or no options to prevent, treat, or cure the disease, a user opt-in section must be provided. This opt-in page must be provided for each disease that falls into this category and must provide specific information relevant to each test result. The opt-in page must include:
(i ) An option to accept or decline to receive this specific test result;
(ii ) Specification of the risk involved if the user is found to have the specific genetic test result;
(iii ) Professional guidelines that recommend when genetic testing for the associated target condition is or is not recommended; and
(iv ) A recommendation to speak with a health care professional, genetic counselor, or equivalent professional before getting the results of the test.
(3 ) Frequently asked questions (FAQ) page: This page must provide information that is specific for each variant/disease pair that is reported. Information provided in this section must be scientifically valid and supported by corresponding publications. The FAQ page must explain the health condition/disease being tested, the purpose of the test, the information the test will and will not provide, the relevance of race and ethnicity on the test results, information about the population to which the variants in the test is most applicable, the meaning of the result(s), other risks factors that contribute to disease, appropriate followup procedures, how the results of the test may affect the user's family, including children, and links to resources that provide additional information.
(M) User comprehension study: Information on a study that assesses comprehension of the test process and results by potential users of the test must be provided.
(1 ) The test manufacturer must provide a genetic risk education module to naive user comprehension study participants prior to their participation in the user comprehension study. The module must define terms that are used in the test reports and explain the significance of genetic risk reports.
(2 ) The test manufacturer must perform pre- and post-test user comprehension studies. The comprehension test questions must include directly evaluating a representative sample of the material being presented to the user as described in paragraph (b)(3)(ii) of this section.
(3 ) The manufacturer must provide a justification from a physician and/or genetic counselor that identifies the appropriate general and variant-specific concepts contained within the material being tested in the user comprehension study to ensure that all relevant concepts are incorporated in the study.
(4 ) The user study must meet the following criteria:
(i ) The study participants must comprise a statistically sufficient sample size and demographically diverse population (determined using methods such as quota-based sampling) that is representative of the intended user population. Furthermore, the study participants must comprise a diverse range of age and educational levels and have no prior experience with the test or its manufacturer. These factors shall be well defined in the inclusion and exclusion criteria.
(ii ) All sources of bias must be predefined and accounted for in the study results with regard to both responders and non-responders.
(iii ) The testing must follow a format where users have limited time to complete the studies (such as an onsite survey format and a one-time visit with a cap on the maximum amount of time that a participant has to complete the tests).
(iv ) Users must be randomly assigned to study arms. Test reports in the user comprehension study given to users must define the target condition being tested and related symptoms, explain the intended use and limitations of the test, explain the relevant ethnicities in regard to the variant tested, explain genetic health risks and relevance to the user's ethnicity, and assess participants' ability to understand the following comprehension concepts: The test's limitations, purpose, appropriate action, test results, and other factors that may have an impact on the test results.
(v ) Study participants must be untrained, be naive to the test subject of the study, and be provided the labeling prior to the start of the user comprehension study.
(vi ) The user comprehension study must meet the predefined primary endpoint criteria, including a minimum of a 90 percent or greater overall comprehension rate (i.e., selection of the correct answer) for each comprehension concept. Other acceptance criteria may be acceptable depending on the concept being tested. Meeting or exceeding this overall comprehension rate demonstrates that the materials presented to the user are adequate for over-the-counter use.
(vii ) The analysis of the user comprehension results must include results regarding reports that are provided for each gene/variant/ethnicity tested, statistical methods used to analyze all data sets, and completion rate, non-responder rate, and reasons for nonresponse/data exclusion. A summary table of comprehension rates regarding comprehension concepts (e.g., purpose of test, test results, test limitations, ethnicity relevance for the test results, etc.) for each study report must be included.
(4) The intended use of the device must not include the following indications for use:
(i) Prenatal testing;
(ii) Determining predisposition for cancer where the result of the test may lead to prophylactic screening, confirmatory procedures, or treatments that may incur morbidity or mortality to the patient;
(iii) Assessing the presence of genetic variants that impact the metabolism, exposure, response, risk of adverse events, dosing, or mechanisms of prescription or over-the-counter medications; or
(iv) Assessing the presence of deterministic autosomal dominant variants.
[82 FR 51561, Nov. 7, 2017, as amended at 83 FR 25914, June 5, 2018]
| 





